Blog
本記事は、2025年夏季インターンシッププログラムで勤務された橘川さんによる寄稿です。
背景
高分子のシミュレーション
高分子とそのシミュレーション
高分子はモノマーと呼ばれる小分子が繰り返し結合した巨大な分子であり、私たちの身の回りの多くの製品に使われています。高分子物性のうち重要なものとして熱特性や機械特性が挙げられます。例えばガラス転移温度 (\(\scriptsize T_g\)) は高分子がガラスのように硬い状態からゴムのように柔らかい状態へ変わり始める温度のことで、一般に\(\scriptsize T_g\)が高いほどそのポリマーをより高温で利用することができます。
このような高分子物性は実際の重合で生じる配列 (モノマーの並び方) や枝分かれ、分子量分布などの構造に強く依存します。そのため、高分子のシミュレーションでは大まかに
- 高分子構造の作成
- 高分子物性の評価
の2段階に分けることができます。しかし既存のシミュレーションでは高分子構造については実際に反応を考慮することは稀で、単純な構造を用いるか経験的知見に基づいて事前に用意することがほとんどです。
高分子反応シミュレータ
現在PFNが三井物産株式会社と共同開発している高分子反応シミュレータ pfpolyでは、
- 重合反応ミュレーションから
- 物性評価シミュレーションまで
を一貫してシミュレートすることで、重合反応シミュレーションによって生成された構造をそのまま物性評価することができます。
高分子のシミュレーションは分子動力学 (Molecular Dynamics; MD) と呼ばれる手法が用いられます。これは簡単にいうとニュートンの運動方程式をコンピュータ上で解くことで分子の動きを追うという手法です。このとき重要となってくるのが運動方程式に出てくる原子にかかる「力」 (ポテンシャル) なのですが、通常のMDでは分子の決まった結合に対して簡単な形を仮定した古典ポテンシャルを使います。
しかし、従来のMD手法では基本的に化学反応を扱うことはできません。これは
- 古典ポテンシャルの精度・表現力が不十分
- 化学反応がレアイベント
であるという2点が課題となります。pfpolyの重合反応シミュレーションでは、前者の課題についてはより精度の高い量子化学計算の結果を学習したニューラルネットワークポテンシャル (NNP) であるPFPを用い、後者については結合の解離や生成にバイアスを加える反応加速MDを行うことでこれらの課題を解決しています。
一方で重合反応シミュレーションを行った後の物性評価シミュレーションの多くは基本的に古典ポテンシャルの精度で十分であり、表現力の高いPFPよりも単純な関数系を持つ古典ポテンシャルを用いる方が高速に動作するためより効率的にシミュレーションすることが可能です。pfpolyはこのようにPFPと古典ポテンシャルの2つのポテンシャルをうまく活用することで反応から物性までの一貫した評価が可能となっています。
ポテンシャルの切り替えに伴う課題
しかし、重合反応シミュレーション (PFP+反応加速MD) から物性評価シミュレーション (古典ポテンシャルMD) に移る際に課題があります。
それは「PFPで反応加速した後の構造にどのように結合をアサインするか」という課題です。
これはポテンシャルの特性の違いによるもので、PFPは元素と座標だけあれば推論が可能なのに対し、古典ポテンシャルは結合に基づいた関数系なので、結合の情報 (結合の有無、ポテンシャルによってはさらに結合の次数) も必要となります。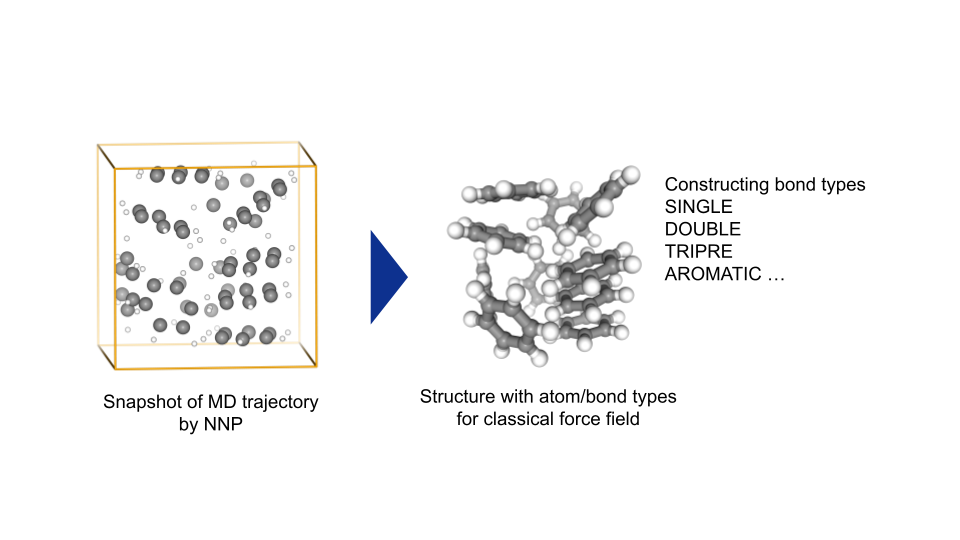
方法
ルールベース結合次数推定モデル[1]
ルールベース手法の詳細
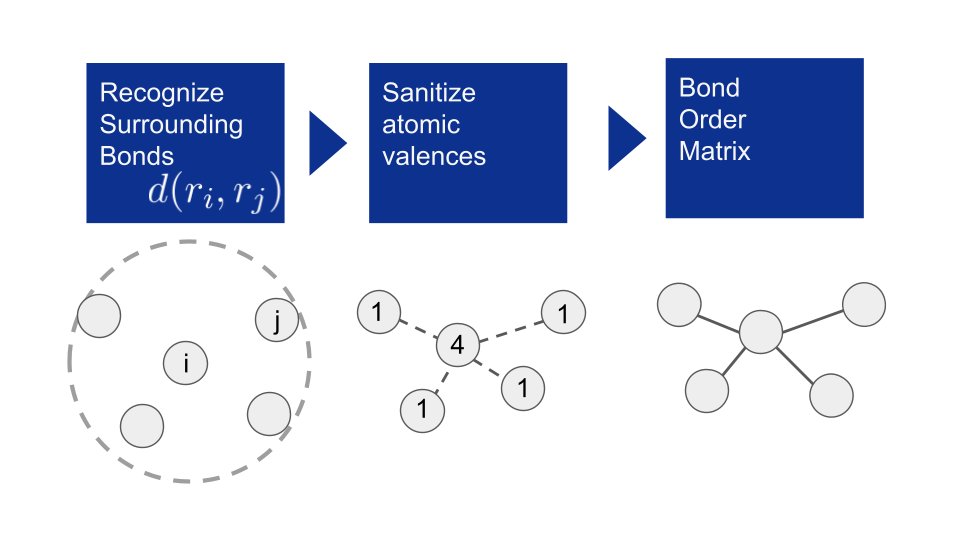
まずは既存の手法であるルールベースの結合次数決定手法について説明します。この方法はRDKitのrdDetermineBondsに実装されており、本研究ではrdDetermineBondsを周期境界条件に対応させたコードを実装して使用しました。ルールベース手法ではまず原子間距離に基づいて、原子種ごとに決められた閾値以内の原子同士を結びます。そしてすべての原子の価数が満たされるように事前に設定された電荷などを考慮しながら結合次数を決定します。ここで重要であることは、閾値以下の結合のみを認識しているということです。
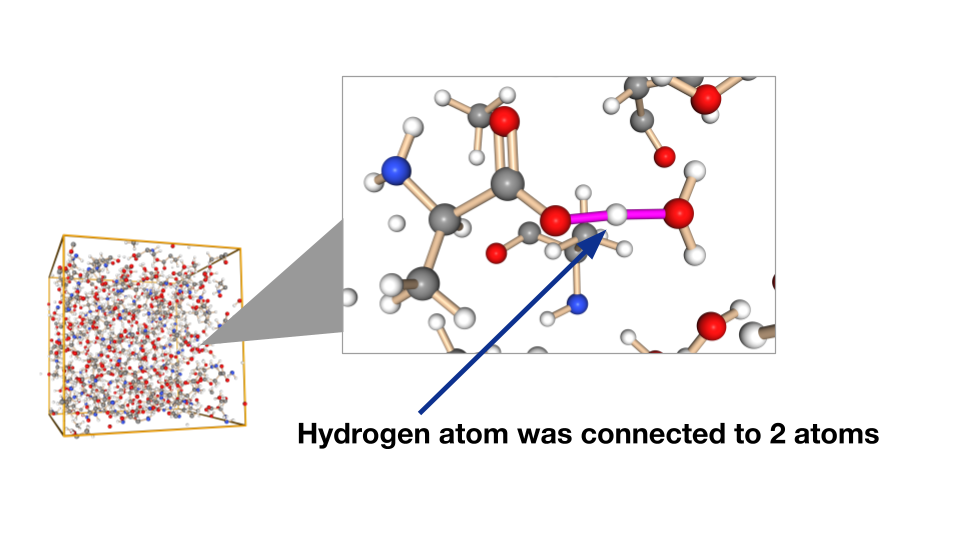
ルールベースで推定できない例
このルールベースの手法は安定な構造 (平衡構造) 付近では結合次数の決定ができるものの、化学反応が起きているような一部が不安定となっている構造の場合に結合次数の推定に失敗することがあります。以下にその一例を示します。図中の構造はアラニン水溶液であり、stickは閾値以内の原子を結んだ結果です。中心に存在するアラニン分子と水分子で水素原子が移動していますが、このような場合に水素原子から水分子の酸素とカルボキシル基の酸素の両方に結合が形成されていることが分かります。水素の価数は中性の場合1であるため、ルールベース手法は整合性のとれた結合次数を決定できず、古典力場に必要な結合の情報を出力することができません。
GNNベース結合次数推定モデルと本研究の目的
本研究のベースとなるモデル[2]
ルールベースではなくグラフニューラルネットワーク (GNN) に基づく結合推定モデルとして、YuelBond が提案されています。このモデルは原子をノード、原子間の関係をエッジとする分子グラフで分子を表現し、各原子のまわりの一定距離 (カットオフ半径) 内にある近傍を局所的な部分グラフとして扱います。隣接ノードやエッジから原子種や原子間距離の情報を集約しながら特徴ベクトルを段階的に更新し、最終的に原子対ごとの結合次数 (結合なし・一重結合・二重結合など) の確率分布を出力し、最も確からしい結合タイプを割り当てます。GNN ベースの利点としては周囲の環境の情報を取り込んだ柔軟な推定が可能であり、ルールベース手法が結合割り当てに失敗する場合でも結果を返すことができます。実際に YuelBond は、平衡構造にノイズを加えた分子の結合次数推定タスクで、代表的なルールベース手法である rdDetermineBonds より高精度であったと報告されています[2]。一方で、このモデルは水素原子を入力に含めていないこと、学習対象が孤立分子に限られていることが制約として挙げられます。
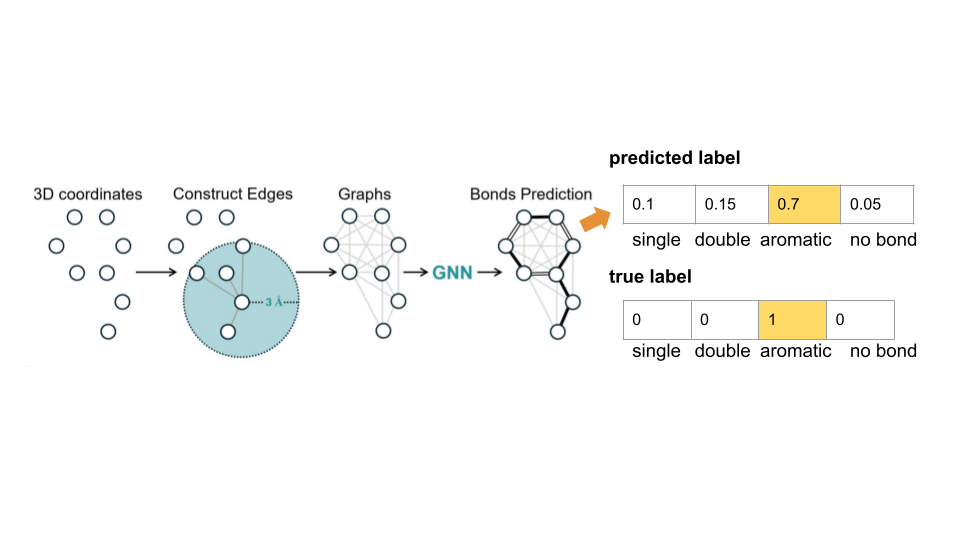 YuelBondの概要 ([2]より引用した図をもとに作成)
YuelBondの概要 ([2]より引用した図をもとに作成)
本研究の方針と目的
そこで本研究ではGNNに基づく凝集系MDトラジェクトリに対する結合次数推定手法を開発します。具体的にはまず先行研究のモデルに対して水素原子をあらわに扱い、MDトラジェクトリのような周期境界条件が適用された構造に対応できるよう拡張しました。次に水素原子をあらわに扱った場合に正確な結合推定が可能であるか、また分子の局所的な構造をもとに巨大分子の結合推定が可能であるか、を確認するために炭化水素分子の孤立分子系のMDデータセットを用いて結合推定を行いました。また高分子重合構造のような凝集系での結合推定が可能であるか確認するため液体構造のMDデータセットを用いて結合推定を行いました。加えて化学反応が起きている中間体構造での結合推定の例として、アラニン水溶液構造に対して本研究モデルで検証を行いました。最後に高分子であるPMMAの分解をシミュレートしたMDトラジェクトリに本研究モデルを適用することで凝集系ダイナミクスへの適用可能性を検証しました。
本研究の学習モデル
本研究の学習モデルは先行研究であるYuelBondの入力に水素原子を追加したものを使用しました。また凝集系のMD計算は通常周期境界条件を課して行われるため、モデルの構造推定とトレーニングデータを周期境界条件に対応させました。
データセット
オリゴマーデータセット
ここでは水素原子を含めた構造として炭化水素のオリゴマーデータセットを作成しました。孤立分子形の計算のために大きな立方体 (\(\scriptsize 100\ \mathrm{Å}^3\)) の中心に一つだけ分子が存在する構造を作成しました。MD計算はPFPを用いてNVTアンサンブル (\(\scriptsize d \lt 0.005\ \mathrm{g/ml}\), \(\scriptsize T=1200\ \mathrm{K}\)) で行い、\(\scriptsize 100\ \mathrm{fs}\)ごとのスナップショットからトレーニングデータを作成しました。高温のMDで結合の解離などがおこらないよう、結合長に調和関数様の拘束を加えました。正解ラベルはSMILESから構造を生成する際に事前に与えた結合次数を正解としました。局所的な情報の拡張性を確認するため、トレーニングデータは重合度\(\scriptsize n=2\mathrm{–}9\)の小分子、テストデータは重合度\(\scriptsize n=20,\ 30\mathrm{–} 90\)の中~大分子を使用しました。
純物質・水溶液の液体構造データセット
ここでは基礎的な有機分子の液体構造を用いてNPTアンサンブル (\(\scriptsize P=1.0 \times 10^5\ \mathrm{Pa}\), \(\scriptsize T=300 \ \mathrm{K}\)) でMD計算を行い、データセットを作成しました。オリゴマーデータセットの場合と同様に、MD計算はPFPを用いて行い、\(\scriptsize 100\ \mathrm{fs}\)ごとのスナップショットからトレーニングデータを作成しました。正解ラベルはSMILESから構造を生成する際に事前に与えた結合次数を正解としました。また高分子の重合構造などの溶媒と溶質が混ざり合っているような状況を想定して、水と上記の有機分子が同数含まれている水溶液の構造でもデータセットを作成しました。
PMMAの分解反応のMDトラジェクトリ
実用的なテストデータとしてポリメタクリル酸メチル (PMMA) の分解反応のMDトラジェクトリに対して結合次数推定を行いました。この構造では結合の解離生成が行われているような箇所が多数存在するため、ラジカル分子が含まれています。
結果・考察
結果の評価方法
本研究では結合次数の推定精度の評価として、結合ごとの正答率 (per-bond accuracy) とスナップショットごとの正答率 (per-snapshot accuracy) を評価しました。正解したスナップショットは「構造中のすべての結合が正しく推定されたスナップショット」と定義されました。液体構造内の結合は10000本以上存在することから、スナップショットごとの正答率は結合ごとの正答率に比べてかなり厳しい指標となります。実際に古典MDへ切り替える際にはすべての結合を正確に決定できている必要があるため、スナップショット単位で結合推定できていることが重要です。それぞれの正答率は以下の式で計算されました。

オリゴマーデータセットの結果
ルールベース手法と本研究モデルの比較
小分子の炭化水素オリゴマーデータセットで訓練したモデルを、中~大分子でテストした結果を表に示します。結合ごとの正答率は本研究モデルとルールベース手法のどちらでも99.99%以上と高精度でした。この結果から本研究モデルは局所的な情報から巨大分子へと一般化が可能であり、水素をあらわに扱った場合でも結合推定を高精度に行うことができることが示されました。またスナップショットごとの正答率ではどちらの手法でも90%程度と低下する結果となりました。これは約10%の構造に少なくとも一つ推定できていない結合が存在することになります。
| per-bond accuracy | per-snapshot accuracy | |
|---|---|---|
| This work | 99.997% | 89.78% |
| Rule-based method | 99.990% | 90.77% |
スナップショットごとの正答率が低下する原因
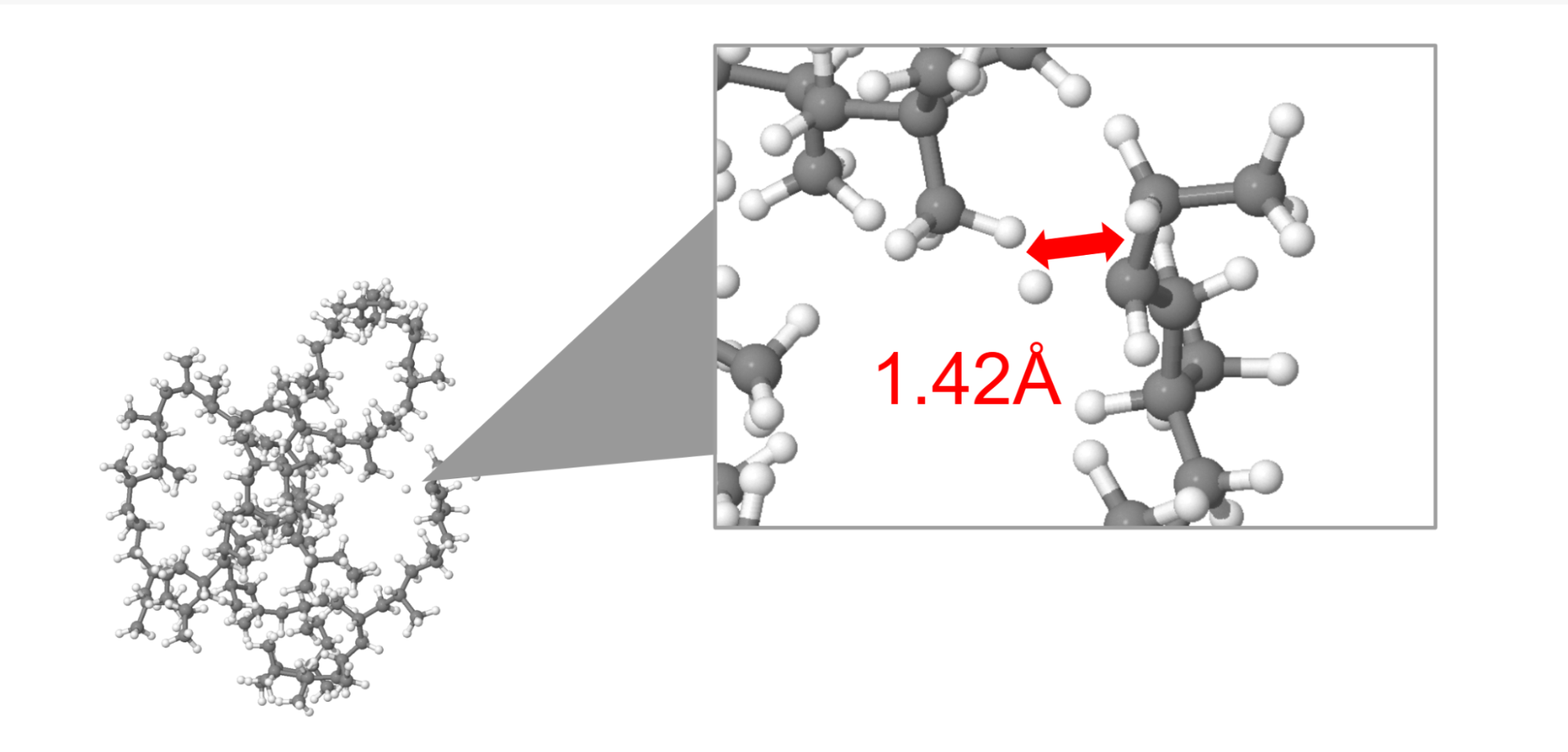
ルールベース手法のスナップショットごとの正答率が低下した原因を調べると、MDデータセット内のC-H結合の結合長が伸びた場合に結合次数の推定が失敗していることがわかりました。赤で示したC-H結合の結合長は\(\scriptsize 1.42\ \mathrm{Å}\)であり、ルールベース手法のC-H結合の閾値である\(\scriptsize 1.39\ \mathrm{Å}\)より長くなっていました。このような場合、ルールベース手法では閾値以上の結合長を持つ結合を認識できないことが原因であると考えられます。GNNベース手法でも同様にトレーニングデータに含まれている構造との差異が大きいために結合推定に失敗していると考えています。
液体構造データセットの結果
ルールベース手法と本研究モデルの比較 (純物質の液体構造)
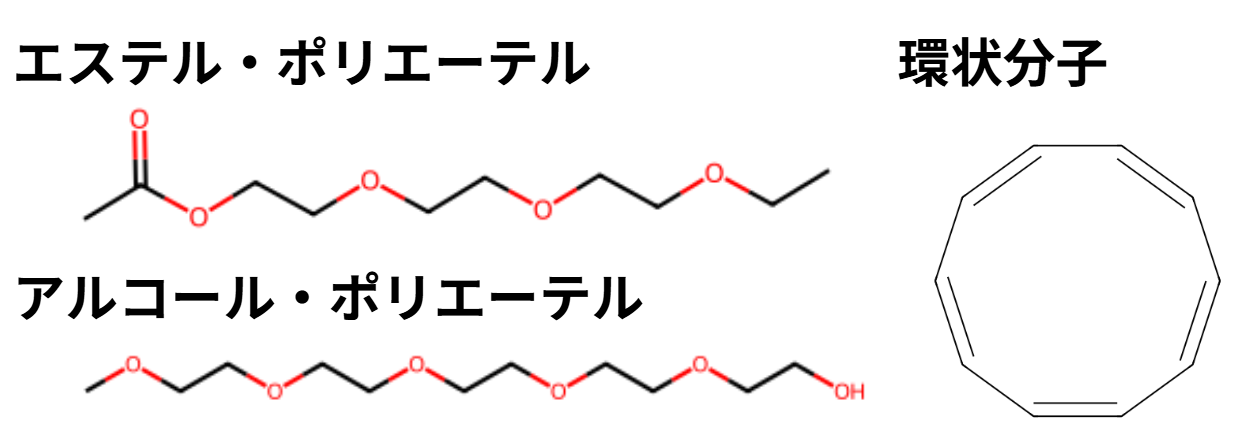
純物質の液体構造データセットでの評価では、トレーニングデータに含まれている分子と類似の分子である、エステル、エーテル、環状分子で推定精度を評価しました。分子ごとの精度を表に示します。エステル、エーテルではルールベース手法と本研究モデルのどちらもほぼ100%で結合推定できているのに対して、環状分子では推定に失敗している場合が見られました。エステル、エーテルの推定結果が100%となった理由としては、常温付近でのMDトラジェクトリであるため構造の揺らぎが小さかったことが理由であると考えています。環状分子についてはまとめて下で考察しています。
| エステル・ポリエーテル | アルコール・ポリエーテル | 環状分子 | ||||
|---|---|---|---|---|---|---|
| per-bond accuracy | per-snapshot accuracy | per-bond accuracy | per-snapshot accuracy | per-bond accuracy | per-snapshot accuracy | |
| This work | 100.00% | 100.00% | 100.00% | 100.00% | 99.993% | 48.00% |
| Rule-based method | 100.00% | 100.00% | 100.00% | 100.00% | 50.00% | 0.00% |
ルールベース手法と本研究モデルの比較 (混合物の液体構造)
混合物の液体構造データセットでの評価では、純物質の液体構造の場合と同様にトレーニングデータに含まれている分子と類似の分子である、エステル、エーテル、環状分子で推定精度を評価しました。混合物の構造は溶質分子と水分子が同数ずつ含まれている構造を作成しました。分子ごとの精度を表に示します。エステル、エーテルではルールベース手法と本研究モデルのどちらもほぼ100%で結合推定できているのに対して、環状分子ではスナップショットごとの正答率でみるとすべての構造で推定に失敗している場合が見られました。
| エステル・ポリエーテル | アルコール・ポリエーテル | 環状分子 | ||||
|---|---|---|---|---|---|---|
| per-bond accuracy | per-snapshot accuracy | per-bond accuracy | per-snapshot accuracy | per-bond accuracy | per-snapshot accuracy | |
| This work | 99.9994% | 92.0% | 100.0% | 100.0% | 99.79% | 0.0% |
| Rule-based method | 100.0% | 100.0% | 100.0% | 100.0% | 54.58% | 0.0% |
結合次数の推定に失敗した例とその原因
環状分子で結合推定に失敗している例としてルールベース手法で推定した結果、環状分子の二重結合がすべて一重結合として判定されているために結合推定に失敗していました。この理由として、環状分子のひずみが大きく、不自然な結合長となっているために誤った部分に結合を生成してしまったり、反対にoligomerデータセットの場合と同様に結合が伸びて結合を認識できていないことが原因と考えられます。また環状分子は\(\scriptsize 4n+2\)電子系であり芳香族性を有していることも、正答率が低下した一因となっている可能性もあります。
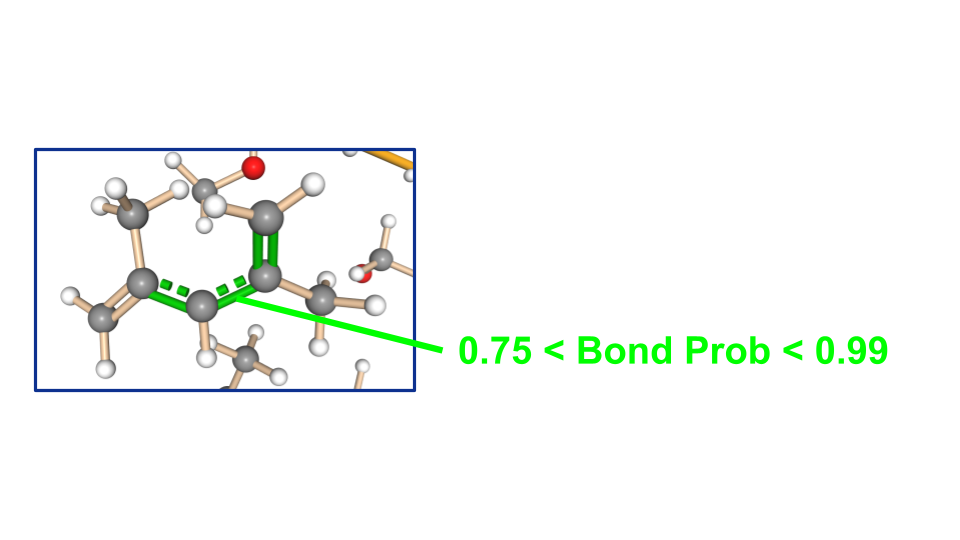
水素結合を形成した構造に適用した結果
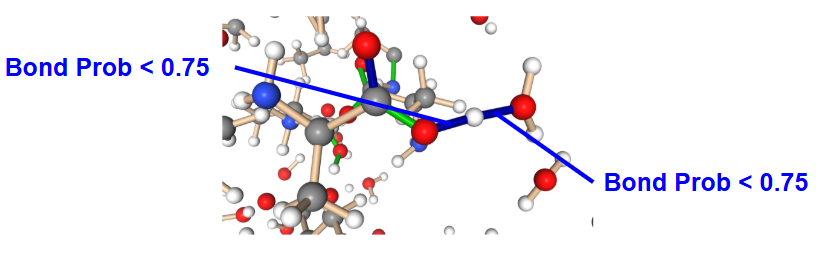
次に化学反応の中間体のような構造に本研究モデルを適用した場合の推定結果を示します。図はアラニン分子と水分子の混合物中でアラニン分子のカルボキシル基同士で水素原子が移動している構造です。本研究モデルで結合推定を行うと、ルールベース手法の場合と同様に両方の酸素と結合していると誤って判定されました。詳しく解析して結合次数の確率分布をみてみると、水素結合の周辺の推定確率が低下していることが分かりました。この図では推定された結合次数の確率が99%以上のものは通常の色で、75~99%のものを緑、75%以下のものを青で示しています。以上のことから原子がどちらに結合しているか判定することが難しい構造の場合には、推定確率も対応して低下していることがわかります。
高分子MDトラジェクトリの結合次数推定
ここでは実用的なタスクとして、結合の解離形成が行われるような構造に対して適用しました。具体的にはポリメタクリル酸メチル (PMMA) の分解反応のMDトラジェクトリについて、ルールベース手法と本研究モデルの両方を適用した結果を示します。
ルールベース手法で推定できない例に適用した結果
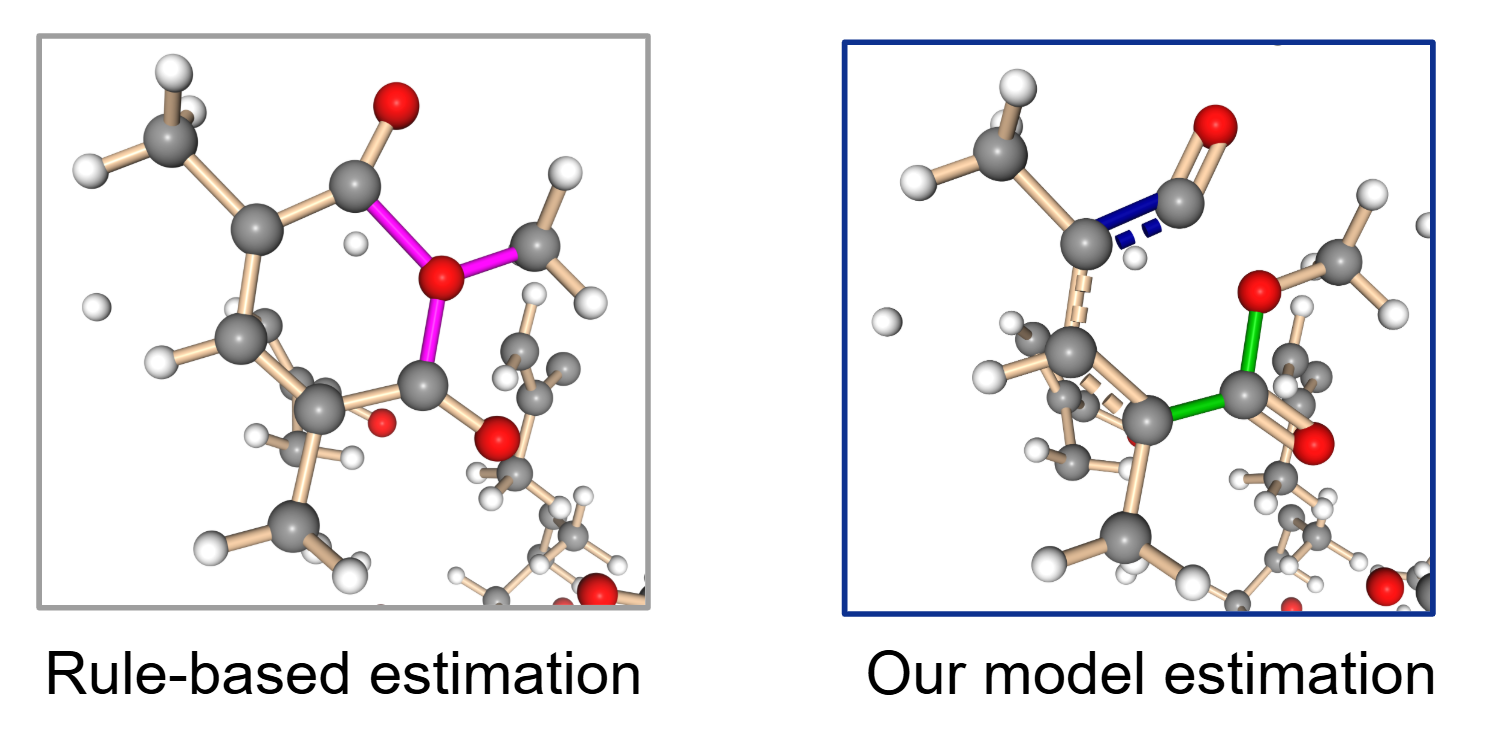
高分子MDトラジェクトリの内、ルールベース手法で結合の推定に失敗した構造に本研究モデルを適用した結果を示します。この図では推定された結合次数の確率が99%以上のものは通常の色で、75~99%のものを緑、75%以下のものを青で示しています。ルールベース手法ではマゼンタで示した部分の酸素の価数が3となっているのに対して、本研究モデルでは酸素の価数が2となっており、価数に従った結合推定となっています。この分子自体が高分子が分解する過程に生じるラジカル分子であるため、分子全体の価数が満たされている結果を得ることはできません。しかし本研究モデルの推定結果 (右) はできるだけ化学的に辻褄が合うような構造を出力していることが分かります。また推定の信頼度に対応して酸素原子の周辺の推定確率が低下していることが分かります。
結合推定に失敗している分子の推定結果
同様に本研究で得られた結合次数の推定結果をもう一例図に示しました。分子全体はラジカル分子となっているため結合次数の決定が難しい構造であるといえます。この図では推定された結合次数の確率が99%以上のものは通常の色で、75~99%のものを緑で示しています。本研究モデルの推定結果では中心に存在するC-C結合を共役結合と判定し、ラジカルであれば正解となる構造を出力していることが分かります。加えてもう少し詳しく観察してこの炭素周辺の確率分布を見ると、周囲の推定確率に比べて低下していることがわかりました。ここから、ラジカル分子や電荷を帯びているなど結合次数を決定することが難しい分子の結合次数推定では、その難しさが推定確率の低下という形で結果に反映されていることがわかります。
考察
以上の結果から、高分子MDトラジェクトリのすべての結合を正確に割り当てることはできないものの、ラジカルになっている分子でも化学的に正しそうな推定結果を出力しているという結果になりました。本研究モデルがルールベース手法のように推定に失敗すると結果が得られないのではなく、推定が難しい構造であっても確率分布の形で結果を得られるというのは古典力場への切り替えの際に有用な情報であると考えています。とはいえ結合次数の決定が難しい構造であっても、NNPから古典力場への切り替えの際には結合次数を割り当てなくてはなりません。今後はそのような難しい分子に対しては、ラジカルや電荷などを考慮した複数の割り当て候補を考えて、分子全体の推定確率が最も大きくなる候補を採用するなどのスキームが考えられます。またMDトラジェクトリは時系列データですので、スナップショットごとに評価するのではなく前後のスナップショットの出力結果を参照しながら結合次数を割り当てていく方針も考えられます。
まとめ・今後の展望
本研究ではGNNに基づく結合次数推定モデルを作成し、オリゴマーデータセットと液体構造のデータセットを用いて訓練しました。データセットと異なる分子で評価したところ、大体の構造ではルールベース手法と本研究モデルのどちらも正確に結合次数を推定できていたものの、ひずみの大きな構造では推定の精度が低下するという結果となりました。また高分子MDトラジェクトリに適用した所、重合反応の中間体のような不安定な構造では結合次数を決定することができていないという結果になりました。決定できていない箇所の推定確率が低下していたことから、結合次数を決定しにくい構造の場合には推定確率の形でそれを表現しているといえます。
今後の改善点としてはモデルのアーキテクチャについて、先行研究ではカットオフ半径が\(\scriptsize 3.0\ \mathrm{Å}\)であり、これは高々2つ先の結合までしかモデルに見えていないことになります。今後はカットオフ半径を広げることによって共役結合など長距離の情報が重要となる結合推定の精度を向上させることができると期待されます。最後に結合次数の決定に関して、複数の構造の中から結合次数の同時確率が最大となる割り当てを採用するなどが考えられるほか、MDトラジェクトリの時系列データを利用した推論[4]なども有望であると思います。
また本研究は出力として結合次数の確率が得られます。これを利用して反対に「結合が存在している確率を増やす (減らす)」勾配の方向へバイアスをかけたようなMDサンプリングなども考えられるのではないかと期待しています。[5]
最後に
本研究ではメンターの宮崎さん、森さんをはじめとしたMaterials Researchチームの方々に非常にお世話になりました。インターン期間中は毎日疑問点などを解決するミーティングを行っていただき研究テーマや方針について逐一相談ができたため、二か月という短い期間ではありましたが、実りのある研究ができたと思っています。
また本インターンでは結合次数の決定という普段の自分の研究内容と少し離れたテーマとなりました。しかしメンターの方々のご指導の元、機械学習モデルの拡張やPFPを用いたMDデータセットの作成など計算化学と機械学習を融合した研究に必要なスキルを身に着けることができ非常に勉強になりました。特に短い期間で研究を進めるという経験はこれからの自分の研究の姿勢にも活用して行きたいと思います。
インターン期間中、PFNに出社すると計算化学だけでなく様々なバックグラウンドを持つ方々がいらっしゃるのはもちろんのこと、皆さんが自分の専門分野以外の幅広い知識や技術を持ち合わせて活発に議論されていることにとても刺激を受けました。自分も幅広い知識を身に着け自分の研究に積極的に活用していこうと思います。最後になりましたが、PFNの皆様二か月間お世話になりました。ありがとうございました!
参考文献
- Kim, Y., & Kim, W. (2015). Universal structure conversion method for organic molecules: From atomic connectivity to three‐dimensional geometry: From atomic connectivity to 3D geometry. Bull. Korean Chem. Soc., 36(7), 1769.
- Wang, J., & Dokholyan, N. (2025). Multimodal Bonds Reconstruction Towards Generative Molecular Design. bioRxiv: 2025.05.06.652517.
- Takamoto, S., Shinagawa, C., Motoki, D., Nakago, K., Li, W., Kurata, I., Watanabe, T., Yayama, Y., Iriguchi, H., Asano, Y., Onodera, T., Ishii, T., Kudo, T., Ono, H., Sawada, R., Ishitani, R., Ong, M., Yamaguchi, T., Kataoka, T., Hayashi, A., Charoenphakdee, N., & Ibuka, T. (2022). Towards universal neural network potential for material discovery applicable to arbitrary combination of 45 elements. Nat. Commun., 13(1), 2991.
- Wang, L.-P., Titov, A., McGibbon, R., Liu, F., Pande, V. S., & Martínez, T. J. (2014). Discovering chemistry with an ab initio nanoreactor. Nature Chemistry, 6(12), 1044.
- Vandenhaute, S., Braeckevelt, T., Dobbelaere, P., Bocus, M., & Van Speybroeck, V. (2024). Rare event sampling using smooth basin classification. arXiv: 2404.03777v3.
