Blog
本記事は、2019年インターンシップで勤務した 阿部健信 さんによる寄稿です。
こんにちは。2019年夏季インターンに参加した東京大学の阿部健信です。「Chainer Chemistryの大規模グラフのタスクへの拡張」というテーマで取り組んだ内容を説明させていただきます。インターン内容のスライドはこちらにアップロードされています。
TLDR;
- Chainer Chemistryで大規模グラフのデータを扱えるようにしました。
- convolution演算を\( O(V^2) \)から\( O(E) \)にしました。
- メモリ使用量も抑えて、PyTorch Geometricでは動かないRedditデータセット(23万頂点, 1100万辺)を16GBのsingle GPU上で学習できるようにしました。
はじめに
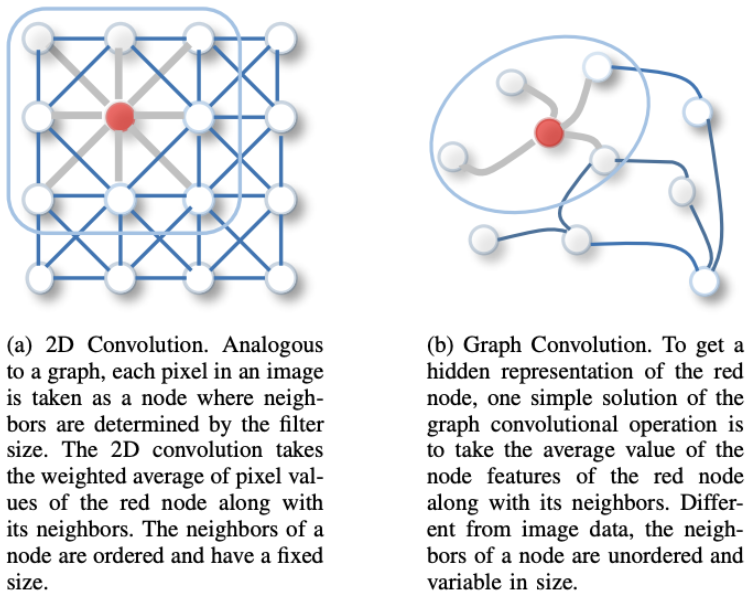
画像に対する2D ConvolutionとGraph Convolutionの比較 [1]
その注目の高まりから、PyTorch Geometric [2]やDeep Graph Library (DGL) [3]といった高機能で最適化されたGNNライブラリの開発が盛んに進められています。
Chainer Chemistryは、PFNが開発しているGNNのオープンソースのライブラリです。
名前からも分かるとおり、もともと分子など化学データへの適用を目的として作られたもので、qm9などの化学データセットが手厚くサポートされています。一方、他にGNNの研究でよく用いられるSNSなどのネットワークデータのサポートはなされていませんでした。
課題内容
今回のインターンのタスクは、Chainer Chemistryでネットワークデータのサポートを行うことです。そのために、大きく以下の2つの内容を行いました。
1. node classificationのサポート
化学分子データなどたくさんの小さなグラフのデータセットに対してGNNは、graph classification/regressionといった、グラフ全体の性質を学習するのに用いられます。
一方、巨大なネットワークデータに対しては、1つのグラフを入力として各頂点ラベルの分類を行うといった異なるタスクに用いられる事が多いです。
[4]で提案されているようなsemi-supervised node classificationへの対応を行いました。
具体的なフレームワークの違いはスライドをご参照ください。
2. 巨大でsparseなグラフのためのGNNの効率的な実装
こちらが今回のインターン内容のメインで、巨大なグラフを動かすためには必要不可欠な内容でした。
以下、\( V \) 個の頂点、\( E \)個の辺からなるグラフを考えます。
Message passingにもとづくGNNでは、各頂点に対して近傍の頂点の特徴量のaggregationの操作を行います。このaggregationの関数はpermutation invariantな様々な関数が用いられ、例えばよく使われるsumの場合は以下の式になります。
\( H’ = AH \)
(\( H \): 頂点の特徴量行列, \( A \): 隣接行列, \( H’ \): aggregateされた特徴量)
既存の実装は全てこの行列演算に基づくものでしたが、これは2つ問題点があります。
1つめは、グラフが疎な際にメモリ的にも実行時間的にも無駄が生じてしまうことです。
2つめは、batch化の際のゼロパディングのオーバーヘッドです。
これらの問題を解決するために、辺の情報を密な隣接行列ではなく、疎なデータ形式で持たせるという事が考えられます。今回のインターンでは、こちらのレポジトリでsparse patternとして紹介されているデータの持ち方を新たに実装しました。
これは辺の情報を\( [2, E] \)のサイズの行列で持つ手法で、PyTorch Geometricでも採用されています。
Sparse patternでは、scatter演算と呼ばれる命令を用いることでaggregation部分の計算量を\( O(E) \)で行うことができます。
またbatch化の際に、複数のグラフを全体として大きな1つのグラフとしてみなすことによってゼロパディングのオーバーヘッド完全になくすことができます。
こちらも、より詳細な手法が知りたい方はスライドをご覧ください。
結果
行列演算による既存実装と、sparse patternによる実装の速度比較は以下のようになりました。
まず、3312頂点、4660辺の疎なネットワークグラフに対しては、CPUでは50倍以上、行列演算との相性が良いGPU上でも2倍以上の速度改善が見られました。
また、1つ予想外だったのは、最大でも38頂点という比較的小さなグラフからなる化学データセットに対してもGPU上でも1.5倍程度の速度改善が見られたことです。
これには、バッチ化のオーバーヘッドをなくす工夫が効いていると考えられます。
sparse patternはグラフのconvolution演算に特化して実装されているため速いもののメモリ使用量にまだ無駄があり、Redditデータセット(23万頂点, 1100万辺)を動かすことはできませんでした。
これについては、ChainerのサポートしているCooMatrix演算によるモデルを用いたところsingle GPU (16GB)で動かすことができました。
これまで触れた、既存の隣接行列・sparse pattern・CooMatrixの3パターンについてまとめると、グラフが疎であったりバッチ化のオーバーヘッドが大きかったりすれば基本的にsparse patternが早く、それではメモリが足りない場合はCooMatrixを使うとよい、という結果になりました。
この結果を踏まえて、3つのパターンを場合に応じて使い分けられるように実装しています。
特に、現状のPyTorch Geometricでは動かすことができないredditなどの超巨大なグラフを動かせるという点は、Chainer Chemistryを使うモチベーションの1つになると思います。
新しいモデルを自分で実装したいときに、ChainerでサポートされているCooMatrix演算を普通の行列演算と同じようなインターフェースで直感的に使えるのも魅力です。
まとめ
今回の成果はChainer Chemistryにマージされています。新しい実装方針に対応しているモデルはまだ多くはありませんが、これからどんどん対応していく予定です。
exampleのコードを動かすことで簡単に巨大グラフ上での学習ができるようになっているので、ぜひ試してみてください。
参考資料
[1] https://arxiv.org/pdf/1901.00596.pdf
[2] https://rlgm.github.io/papers/2.pdf
[3] https://rlgm.github.io/papers/49.pdf
[4] https://arxiv.org/pdf/1609.02907.pdf
