Blog
はじめに
こんにちは、Preferred Networksの森穂高です。普段はMaterials&DrugDiscovery事業本部のマテリアルリサーチグループでエンジニアリングマネージャをやっています。
この記事では、先日出版された私たちの論文「Ready-to-Use Polymerization Simulations: Combining Universal Machine Learning Interatomic Potential with Time-Dependent Bond Boosting for Polymer and Interface Design」で提案した、重合・硬化反応を扱う分子動力学(MD)の枠組みを紹介します。
ポリマー材料は、最終的にできあがる分子構造(鎖長、分岐、架橋、界面での結合など)で物性が大きく変わります。実験では反応の進行や平均的な構造は観測できても、反応中間体や界面近傍での結合再編成を原子レベルで追い切るのは難しいことが多いです。こういったシチュエーションで計算化学の活用が考えられますが、反応を含むMDを現実的なコストで回すのはまだ手強い、というのが現状です。
今回の論文では反応を含むMDを「とりあえず回して、機構と相対傾向を見にいける」状態まで持っていくための手法です。絶対の速度定数を当てにいくというより、材料設計で必要になる比較や探索に使えるところを狙っています。この論文のコンセプトムービーがyoutubeに公開されていますのでこちらもご覧ください。
重合・硬化のMDが手強い理由
重合・硬化をMDで扱うときの難しさは、大きく2つあります。
1つ目は、反応を扱えるポテンシャルの準備が重いことです。ReaxFFのような反応力場は便利ですが、対象系や反応クラスが変わると、パラメータセットの選定や妥当性確認が必要になります。特に接着や摩擦などの実用的なタスクで頻繁に登場するで有機-無機界面では、有機物の反応や分子の凝縮構造、無機物の結晶構造系と幅広い物理化学的性質を両立できるポテンシャルが必要であり、こういったポテンシャルを調査・検証し、利用可能な段階まで持っていくのに時間がかかりがちです。
2つ目は、反応がレアイベントで、素朴なMDでは反応が進まないことです。一般に反応の発生頻度は活性化エネルギーに対し指数的に減少していきます。例えばラジカルカップリングなどは非常に早い反応として知られていますが、このような反応であっても発生頻度はナノ秒オーダーであり、全原子シミュレーションの典型的な時間刻みであるフェムト秒と比べると、100万ステップが必要となるレアイベントとなります。重合は逐次反応の連続なので、ナノ秒程度のMDでは反応イベントが十分に起きず、統計を取る前に計算が終わってしまうことが多いです。反応を起こすために加速法を入れると今度は、反応ごとにパラメータを調整する手間が出たり、正逆両方向の反応が加速されてしまい重合のような一方向プロセスと相性が悪かったりします。
1つ目のポテンシャルに関しては、ここ数年でDFTデータで学習した汎用機械学習ポテンシャル(uMLIP)が現実的な選択肢になってきました。Matlantisで提供されているPFP、M3GNet、CHGNet、MACEなどが代表例です。これらは「系ごとに力場を作り込まない」という意味で非常に魅力的ですが、uMLIPに置き換えただけではレアイベントの問題は解決しません。そこで今回の論文では、uMLIPの汎用性と、反応を起こすための加速を一体で扱うことにしました。
Time-Dependent Bond Boostという割り切り
基本方針はuMLIPとしてPFPを使い、反応が進まない部分はbond-boostで促進する、というものです。ここで従来のbond-boostと変えたのがバイアスの深さを固定しない点です。Vashisthらの先行研究ではたとえば次のような形で深さ \( f_1 \) が固定されています。
$$
V(r) = f_1{1-\exp[-f_2(r-r_0)^2]}
$$
ここで、\( V(r) \) は対象原子対の距離 \( r \) に応じて付加するバイアスポテンシャル(エネルギー)で、\( r \) は対象原子対の原子間距離です。\( f_1 \) はバイアスの振幅(エネルギー次元を持ち、バイアスの強さ=最大の寄与スケールを規定する)、\( f_2 \) は距離依存性の幅を決める正のパラメータ、\( r_0 \) は距離の基準(閾値)です。ただ、反応の障壁は反応タイプごとに違うので、適切な \( f_1 \) を事前に見積もって調整する流れになりやすく、反応種類が増えるほど準備が面倒になります。そこで我々はバイアスの振幅を時間依存にして、反応が起きない間は単調に大きくしていきます。このような時間依存のバイアスポテンシャルをTime-Dependent Bond Boost (TDBB)とよんでいます。論文では
$$
f_1(t)=\min(\gamma t, f_1^{\max})
$$
の形にして、初期は弱く、反応が起きなければ徐々に強くして「そのうち起こす」設計にしました。ここで \( t \) はシミュレーション時間、\( f_1(t) \) は時刻 \( t \) におけるバイアス振幅、\( \gamma \) は振幅の増加率、\( f_1^{\max} \) は振幅の上限値です。\( \min(\cdot,\cdot) \) は2つの引数のうち小さい方を返す関数です。固定深さを当てにいくのではなく、時間方向に自動でスキャンするという感覚です。もう一つ重要なのは、結合形成と結合解離で別の関数形を使い、生成物側(あるいは解離側)に到達したらbiasが消えるようにしたことです。結合形成側は
$$
V_f(r,t) = f_1(t){1-\exp[-f_2(r-r_0)^2]}
$$
結合解離側は
$$
V_d(r,t) = f_1(t)\exp[-f_2 r^2]
$$
を使っています。ここで \( V_f(r,t) \) は結合形成を促進するための時間依存バイアスポテンシャル、\( V_d(r,t) \) は結合解離を促進するための時間依存バイアスポテンシャルです。いずれも距離 \( r \) と時間 \( t \) に依存し、振幅は \( f_1(t) \) で与えられます。こうすることで、反応が起きた後にもバイアスが残り、結合を解離し続ける状況を避けています。距離の閾値 \( r_0 \) は、反応ごとに設計し直すのではなく、対象原子のvdW半径の和にスケール因子 \( \lambda \) を掛けた
$$
r_0 = \lambda \sum_{a\in{\text{pair}}} r_a^{\mathrm{vdw}}
$$
で決めています。ここで \( r_a^{\mathrm{vdw}} \) は原子 (a) のvdW半径(van der Waals半径)で、上付きの \( \mathrm{vdw} \) はvdW半径であることを示すラベルです。\( \lambda \) は無次元のスケール因子、\( \sum_{a\in{\text{pair}}} \) は対象原子対 \( \text{pair} \) を構成する2つの原子 (a) についての和を表します。ここも「反応ごとの手作業を減らす」ための工夫です。この方法の立ち位置は割り切っています。バイアスは時間とともに大きくなり、最終的に障壁を超えることもあり得るので、hyperdynamicsのような物理時間の再構成(time rescaling)は前提にしません。したがって、この枠組みは絶対速度定数を当てる用途ではなく、機構理解と相対比較を主目的にしています。論文中でも、その理由(time rescalingの成立条件が一般に満たされないこと)を明示しています。
実装とシミュレーションフロー
uMLIPにはPFP v6.0.0(CRYSTAL PLUS D3 mode)を使い、MDエンジンはOpenMM 8.1.1を使いました。バイアスポテンシャルはOpenMM-TorchでTorchScriptとして組み込み、力はPyTorchの自動微分で評価しています。このように自動微分可能な形でエネルギーを記述することでオリジナルのバイアスポテンシャルを実装しやすくしています。自動微分で力を求める実装のパフォーマンスは解析微分による実装より一般に低速ですが、ポテンシャルとしてuMLIPを用いる場合はほとんどのケースでそちらがボトルネックになるため、今回の用途ではバイアスポテンシャルがパフォーマンスに与える影響は軽微です。実際のシミュレーションフローを図1にまとめています。最初に官能基に基づいて反応性原子のグループを定義し、距離条件で反応候補の組を作り、原子が重ならない候補だけを選んでバイアスポテンシャルを付与します。バイアス付きMDで反応を誘起した後はバイアスなしで緩和し、結合状態の変化を判定して反応性グループを更新し、次の反応へ進みます。結合判定はvdW半径に基づくルールを採用し、距離がvdW半径和の60%未満になったら結合とみなす、という形に統一しています。
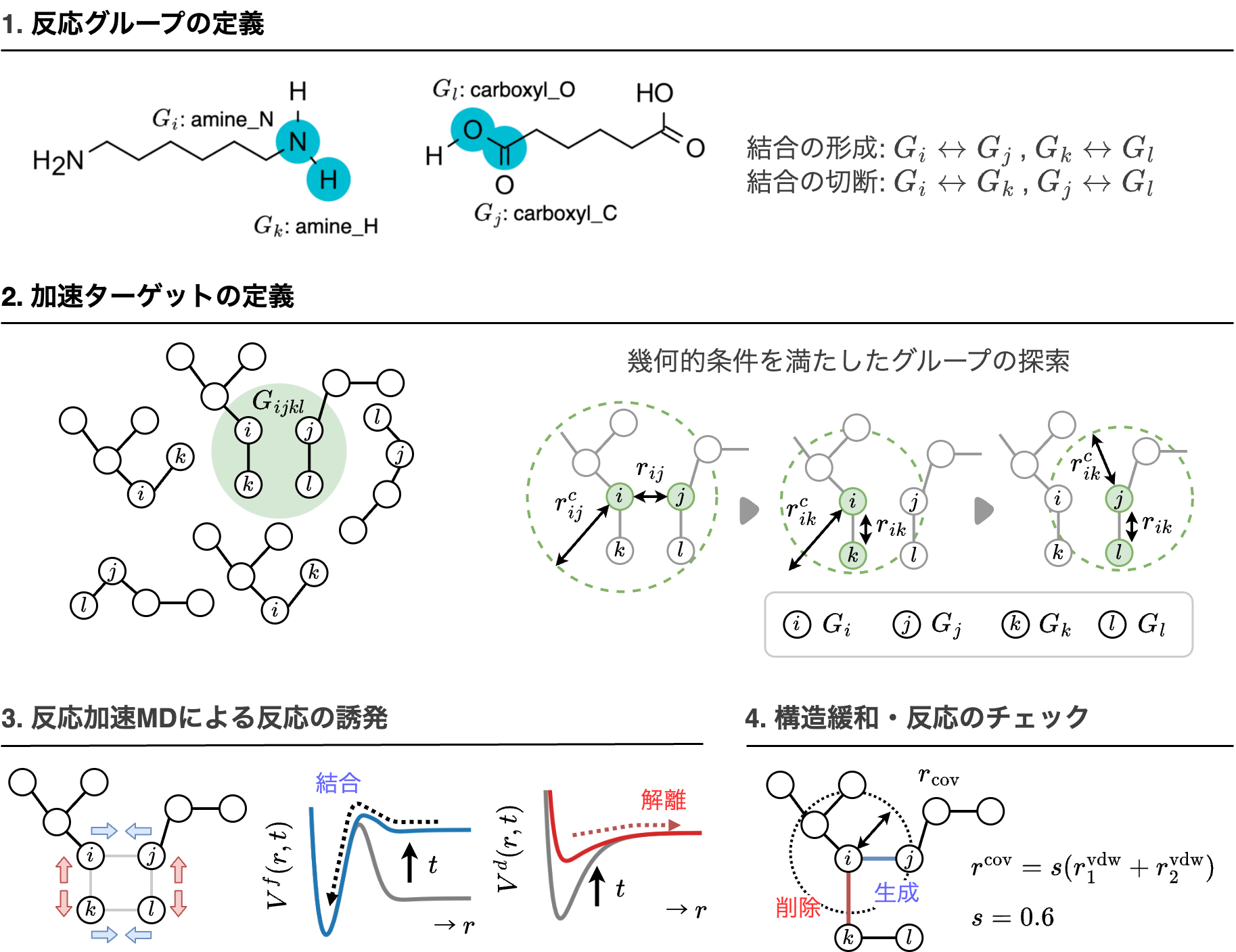
図1 シミュレーションフロー
ケーススタディ
この枠組みは、特定の一反応を精密に当てるより、化学的に違う系を同じ手順で回して、傾向と機構が取れるかを重視しています。そこで論文では、ラジカル重合、縮合重合、界面でのエポキシ硬化の3系で検証しました。ここではその中でも代表的な例として2つを紹介します。
連鎖重合:ラジカル重合における濃度依存と分子量成長のトレンド
複数のビニルモノマー(methyl acrylate、methyl methacrylate、styrene、vinyl acetate、diphenylethylene、dimethyl itaconate)について、モノマー転化率の時間変化を単一指数でフィットして見かけの伝播速度 \(k_p^*\) を取り、実験のランキングと比較しています。結果として、Spearman ρ=0.66、Kendall τ=0.60、rankのMAD=1.0という相関が得られています。完全一致ではありませんが、チューニングなしで相対傾向がある程度出る、という意味では狙いに沿った結果だと思っています。一方で、PFPが推定する障壁順位と実験傾向が一致しない例(styreneの順位など)も出ます。ここは正直に弱いところで、凝縮相では活性化障壁だけでなく拡散や配座、エントロピー要因も効くため、障壁誤差と (k_p^*) の誤差が一対一で対応しない、という点も含めて議論しています。そもそも時間依存バイアスで時間軸が歪むので、絶対速度を当てにいく設計ではなく、ランキングや比較に寄せる、という立ち位置になります。
図2 様々なビニルモノマーに対する見かけの反応速度定数(a)と実験値との序列比較(b)
固体界面での反応:CuO界面でのエポキシ硬化における界面近傍の反応抑制とCu–O–C結合
最後に、CuO(OH終端)基板上でのエポキシ–アミン硬化を扱っています(図3)。この系は、有機硬化反応と酸化物表面反応が同居するため、反応力場でバランスを取るのが難しい類の問題だと思っています。TDBB+PFPはこういった問題に対応できます。シミュレーショントラジェクトリを解析して得られた深さ方向の反応密度(図3 (a))を見ると、界面近傍はエポキシ・アミンの密度が高いにもかかわらず反応密度が下がります。界面近傍では運動性が落ちて反応が抑制される、という理解と整合する結果です。スナップショットでも、CuO–OH表面での開環を介して、ポリマーネットワークと基板の共有結合(Cu–O–Cを含む結合)が形成される様子が確認できます(図3 (b))。数は多くありませんが、界面接着に効く結合が出る、という意味で重要です。
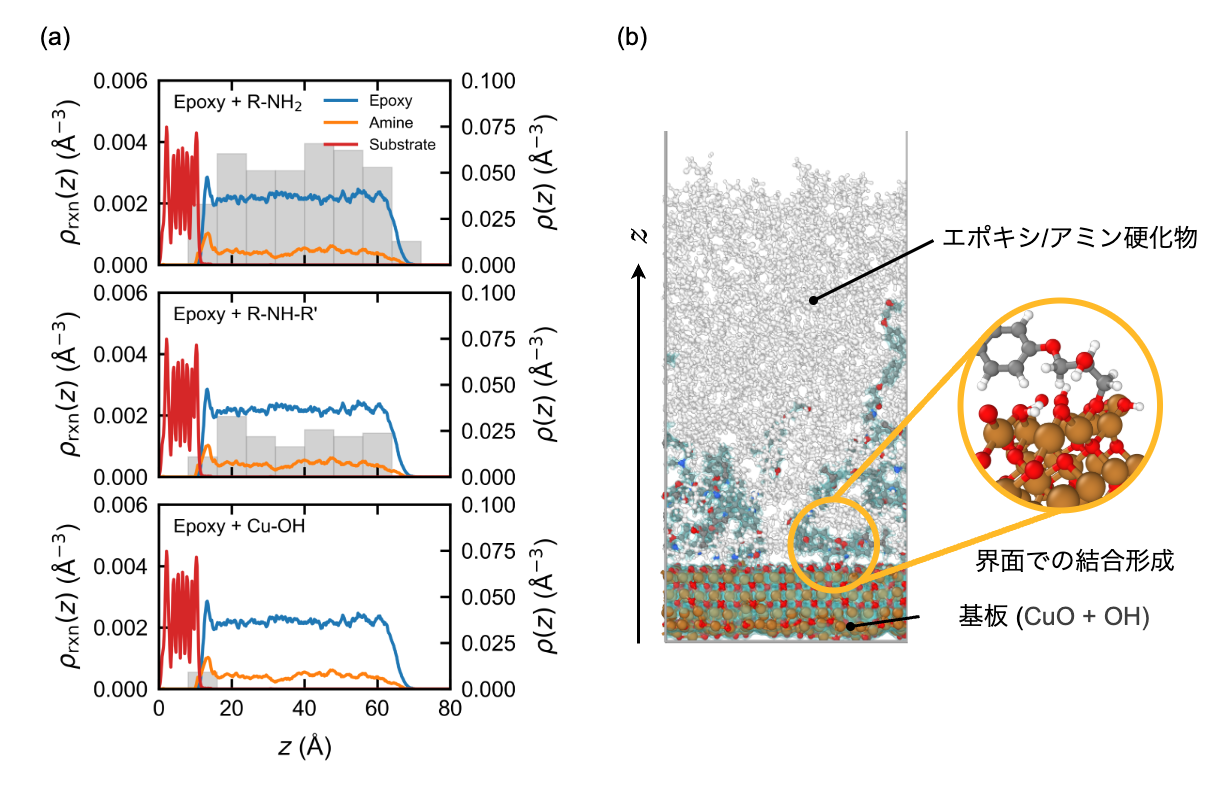
図3 CuO界面でのエポキシ硬化における反応密度プロファイル(a)とスナップショット(b)
手法上の制約と今後の課題
TDBBは「回すこと」に寄せた設計なので、代償もあります。まず、時間依存なバイアスポテンシャルによって物理時間との対応が崩れるため、絶対速度定数を再現する用途には向きません。次に、ラジカル重合については現状の実装では終止・連鎖移動を入れていないので、分子量分布まで含めて再現したい場合は反応カタログの拡張が必要です。最後に、相対反応性の細かい順位付けはuMLIPの反応障壁再現性に上限があり、数kcal/molレベルの差を安定に見分けるのは現状では簡単ではありません。
次の方向性としては、遷移状態や反応障壁の情報をより明示的に含むデータでuMLIP側の障壁忠実度を上げつつ、副反応や終止も含める形で反応カタログを広げ、必要なら加速と時間再構成/kinetic reconstructionを組み合わせて、傾向を見るところから定量に近づけていくのが素直だと考えています。
おわりに
今回の研究は、反応力場を系ごとに作り込む方向というより、uMLIPを前提にして「反応が進まない」というボトルネックだけを、できるだけ手離れよく解く方向を狙いました。TDBBは、物理時間の正確さを捨てる代わりに、反応ごとの事前チューニングをできるだけ避けて、機構と相対比較を取りにいけるようにします。重合・硬化を材料設計のワークフローに載せる上では、まず「回って、見える」ことが重要だと思っていて、その最低ラインを越えるための提案がPFPとTDBBの組み合わせです。
Material &Drug dicoveryは、Matlantis以外にも材料開発を加速させる様々な技術の研究を行っています。一緒に研究開発を行うリサーチャー・エンジニアを募集しています。本記事を通して、ご興味を持っていただけたという方はぜひ下記ページをご確認ください。本記事のような原子シミュレーションの手法に関する知識を持っている方も歓迎です!
