Blog
2026.01.27
創薬DMTAサイクルの高速化! Active Learning駆動型Relative Binding FEP(RBFEP)による”実践的”リード化合物最適化の新戦略
Tag
Yunoshin Tamura
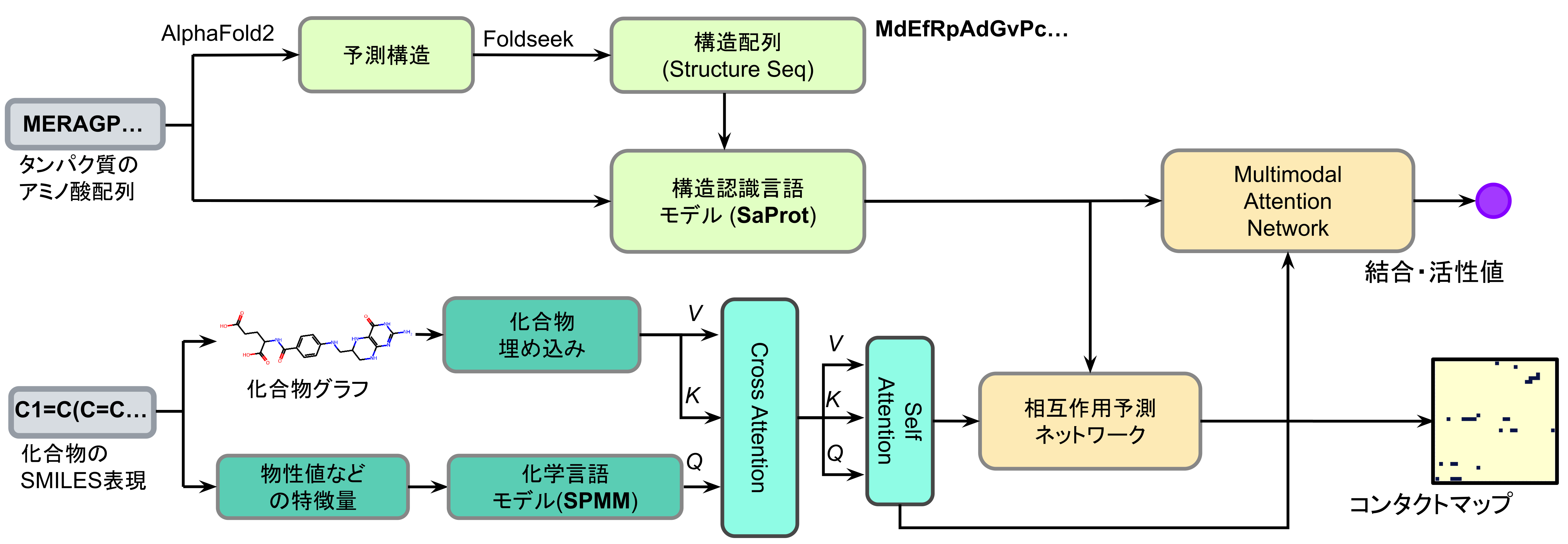
タンパク質に低分子化合物が結合する強度 (Affinity )を高精度に予測する手法として、Free Energy Perturbation (FEP)があります。Preferred Networksは、このFEPに基づき独自に開発した高精度予測技術「P-FEP」を提供しています。しかし、実際の創薬プロジェクトで取り扱うべき化合物数を対象とし、現実的な時間でこの高精度予測を実施するには速度面での課題がありました。そこで、本記事では効率的に大規模化合物群を対象とした高精度予測計算を実現する目的で、機械学習法のひとつであるActive Learning(AL)と統合した「AL-FEP創薬ワークフロー」について紹介します。
はじめに
創薬DMTAサイクル
低分子創薬の初期段階からリード最適化に至るまでのプロセスでは、探索すべき化合物空間(Chemical Space)が極めて広大です。この広大な化合物空間の中から、標的となるタンパク質に対して所望の活性および選択性を示す化合物を効率的に特定することが、創薬研究の成否を左右します。一般に、この最適化プロセスはDesign(設計)、Make(合成)、Test(評価)、Analyze(解析)の4段階を繰り返す「DMTAサイクル」として進行します。膨大な化合物空間から最適解を見つけ出すためには、このDMTAサイクルを高速かつ高精度に回すことが求められます。
特に、時間とコストのかかる「合成・評価(Make/Test)」の工程へ進む前に、いかに「設計(Design)」の段階で有望な化合物を絞り込めるかが重要です。そのため、結合親和性の高精度予測に基づく化合物デザインの優先順位付けは、創薬パイプラインの効率化に不可欠な要素といえます。この課題に対するアプローチとして、タンパク質の立体構造情報を基にしたSBDD (Structure-Based Drug Design )が広く用いられています。
P-FEPとActive Learning
Preferred Networks (PFN )が提供する高精度結合活性予測サービス「P-FEP」[1]は、Relative Binding Free Energy Perturbation (RBFEP )[2]と呼ばれるSBDD技術を基盤とし、共通の分子骨格を持つ化合物群について、タンパク質と化合物の結合に伴う自由エネルギー変化 (結合自由エネルギー\(\Delta G_{\text{bind}}\))を高精度に計算します。P-FEPは様々なタンパク質系において\(\Delta G_{\text{bind}}\)を1.0 kcal/mol以下の誤差で予測できることが確認されています。
本記事では、P-FEPの持つ高精度な予測能力を最大限に活かし、ターゲットとなる化合物空間から、有望な化合物を自動的かつ効率的に探索するために、Active Learning (AL、能動学習 ) と組み合わせた手法「AL-FEP」を紹介します。AL-FEPは、RBFEPによる結合自由エネルギー\(\Delta G_{\text{bind}}\)評価 (ラベル付け )と、機械学習モデルによる次のデータ選択を反復的に繰り返すことで、効率的に化合物空間をガイドする手法です。この方法により、結合自由エネルギー \(\Delta G_{\text{bind}}\)に基づいた化合物デザインの優先順位付けを大規模な化合物群に適用することが可能になり、リード化合物の最適化に至る創薬プロセスを強力に支援します。
方法
1. AL-FEPの概要
AL-FEPワークフローにおいて、機械学習モデルは次に評価すべき化合物を選定する役割を担います。FEP計算とALを組み合わせた先行研究 (TYK2、USP7、D2R、Mproのベンチマークを含む )[3][4]では、機械学習モデルとしてGaussian Process Regression (GPR) モデルの高い性能が示されています。 GPRはベイズ的なアプローチであり、予測値だけでなく、その予測の不確かさ (誤差 )を直接推定できるという重要な特徴を持っています。この不確かさの推定は、後述の「探索 (Exploration)」戦略を実行するために不可欠です。機械学習モデルの入力となる分子の特徴量としては、Morgan Fingerprints (FP)を使用しました。
本記事の手法AL-FEPは、以下の3つの主要ステップを反復的に繰り返すことで、効率的な探索を実現します(図1)。
- 化合物選択: 学習モデルを利用し、化合物プールの中からRBFEP計算を実行すべき化合物を選抜します。計算済みの化合物は選択対象から除外されます。初期状態(全て未計算で学習モデルがない状態)ではランダムまたは化合物空間的に偏りが無いように選択します。
- \(\Delta G_{\text{bind}}\)評価: 選択された化合物に対してRBFEP計算を実行し、高精度な\(\Delta G_{\text{bind}}\)値 (ラベル )を取得します。
- モデル作成/更新: 結合自由エネルギー\(\Delta G_{\text{bind}}\)が計算済み (ラベル付けされた )の化合物セットを用いて、機械学習モデルを作成または更新します。
このサイクルを繰り返すことで、化合物デザインの優先順位付けが\(\Delta G_{\text{bind}}\)に基づいて逐次的に行われ、対象となる化合物空間から有望な化合物を効率的に探索していきます。
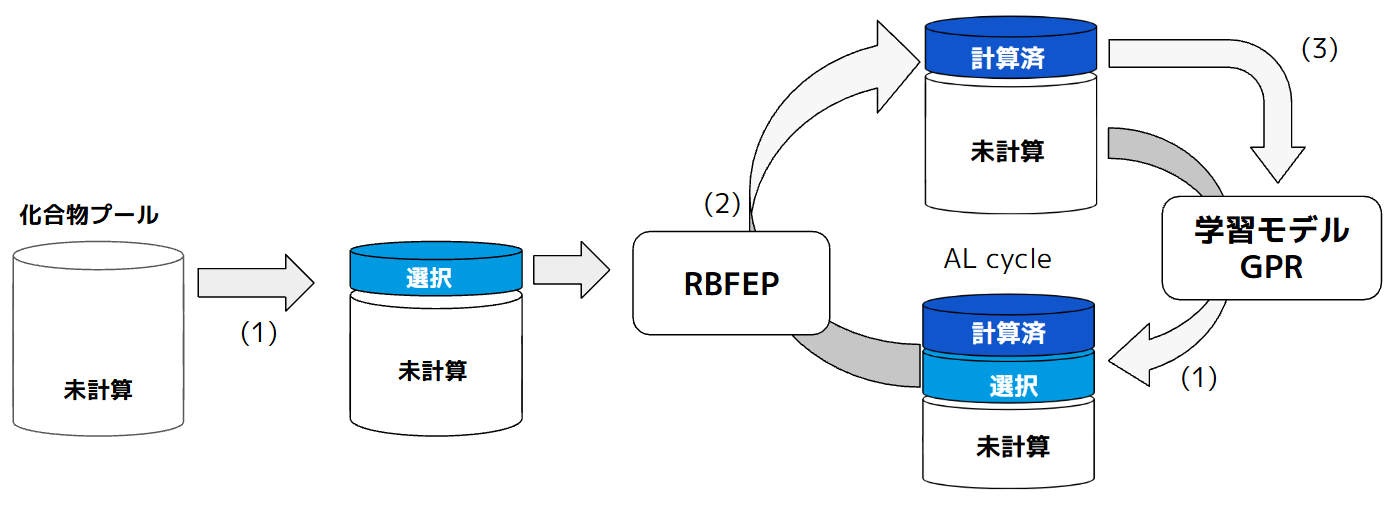
図1. AL-FEPサイクル
2. 計算対象
ターゲット
YTHDC1(YTH N6-methyladenosine RNA binding protein C1)について本手法を適用しました。YTHDC1はm6A (N6-methyladenosine )を認識するリーダータンパク質であり、様々なガンに関わる遺伝子発現の制御に関与しています。標的とする化合物は、SAR (構造活性相関 )情報およびYTHDC1との複合体結晶構造が報告[5]されているピラゾロピリミジン骨格を持つ化合物です。タンパク質の立体構造データには、公開されているPDB ID: 8Q4Tを使用しました。
化合物プール
AL-FEPではPool-based Active Learningを採用しています。様々な方法で作成された化合物プール(コンピューター上に構築した化合物ライブラリ)を利用することができます。たとえば、試薬・化学反応ベースの手法で生成した化合物プールは高い合成可能性が期待できます。一方、機械学習ベースの手法で生成した化合物プールは、試薬・化学反応ベースの手法ではアクセスできないより広範な化合物空間から、多数の化合物の生成が期待されます。
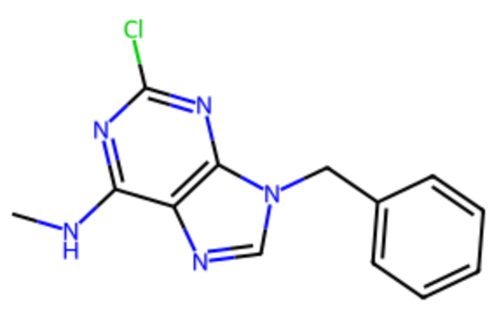
図2. Compound 5の構造
Compound 5 (YTHDC1 IC50 3.0 μM, 図2 )[5]をリファレンスとし、試薬・化学反応ベース(Enamine REAL FragmentおよびBuilding Blocksを試薬情報として使用)および機械学習ベース (REINVENT 4 [6])の手法を用いて化合物プールを構築しました。REINVENTは強化学習 (RL )を用いて、特定の構造的制約(ユーザー定義のスコアリング関数)を満たす新規分子をデザインする生成AIフレームワークです。本検討ではR-グループ置換やスキャフォールド修飾に特化したLibINVENTモードを利用して化合物を生成しました。本事例では化合物生成後に、化合物ディスクリプターベースのフィルター(分子量、原子数、回転可能結合数および脂溶性のような物性値など)と部分構造ベースのフィルターを複数適用し、最終的に表1のような内訳の化合物プールを構築しました。
表1. 化合物プールの内訳
| Data source | Compounds |
| Enamine (Benzyl Halides) | 1,711 |
| REINVENT (LibINVENT mode) | 15,557 |
合成可能性と構造新規性はときにトレードオフの関係にあり、その許容度はプロジェクトによって異なります。本手法を実際のプロジェクトに適用する際は、そのプロジェクトの方針に沿ったフィルターを適用して、化合物プールの品質を担保した上で、本計算に移る必要があります。
3.選択戦略
AL-FEPでは、学習モデルの不確かさと予測値の情報を用いて、探索 (Exploration) と活用 (Exploitation) のバランスを取る化合物選択戦略を採用します。本記事の検討では、Random、 Exploration、 Exploitationを組み合わせた戦略を紹介します。
表2 化合物の選択方法の実装例
| 選択方法 | |
| Random | 化合物プールからランダムに選択します。主に初期バッチの選択用。 |
| Diverse | 多様な化合物を選択し、化合物空間を広くカバーすることを目的とします。主に初期バッチの選択用。 |
| Exploration | GPR(Gaussian Process Regression)で予測された不確かさ (誤差 )が大きいものを選択します。これは、モデルがまだ学習できていない化学空間を広くサンプリングすることを目的とします。 |
| Exploitation | GPRで予測された \(\Delta G_{\text{bind}}\)の良いもの (結合活性が高いもの )を積極的に選択します。 |
4. RBFEPによる結合自由エネルギー計算
RBFEPにおける計算対象化合物ペアの選び方は、化合物の変換経路が閉じるように選ぶclosedマップによるものと、リファレンスとなる化合物に対して結合自由エネルギー差を計算するstar型マップによるものがあります。本取り組みではstar型マップを採用し、計算対象化合物とCompound 5のペアに対して、RBFEPを実行し結合自由エネルギー\(\Delta G_{\text{bind}}\)を算出しました。
結果
構築された化合物プールの約17,000化合物 (表1参照 )の中から、ALサイクルを通じて合計360化合物の \(\Delta G_{\text{bind}}\)をP-FEP(RBFEP)で評価しました。この360化合物は、化合物空間全体のわずか2%に相当します。
1. 選択戦略に基づく化合物空間の探索
本検討では、合計360化合物を、以下の戦略を適用した11サイクルのAL-FEPワークフローで選択しました。
- Random: 60化合物 × 1サイクル (初期バッチ )
- Exploration: 30化合物 × 2サイクル
- Exploitation: 30化合物 × 8サイクル
各サイクルでは、前述の手順 (化合物の選択、RBFEPの実行、GPRモデルの更新 )を繰り返します。各サイクルで得られた化合物の\(\Delta G_{\text{bind}}\)分布を図3に示しました。
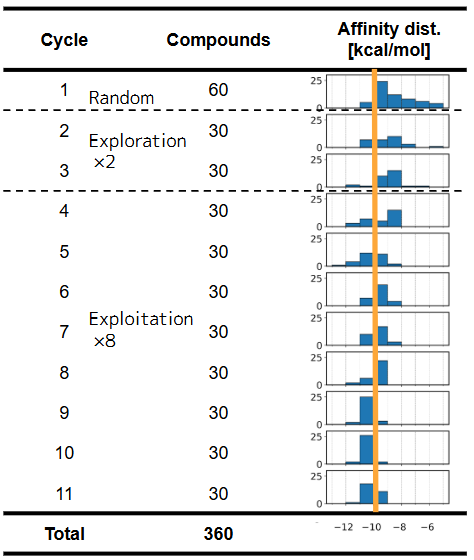
図 3. AL-FEP 各サイクルの結合自由エネルギー\(\Delta G_{\text{bind}}\)分布。\(\Delta G_{\text{bind}}\)は小さいほど(ヒストグラムの左側ほど)結合が強いことを示す。
Exploitation戦略を繰り返すことで、より結合の強い (\(\Delta G_{\text{bind}}\)が小さい )化合物群へと濃縮されていく様子が確認できました。また、化合物プールの化合物空間プロット (図4 )からも、AL-FEPによって選択された化合物が特定の構造クラスターに収束していく様子が確認できました (図4、 図5 )。以上から、AL-FEPが選択戦略に基づいた化合物探索を遂行し、結合が強いと予測される活性化合物群を効率的に取得可能であることがわかりました。
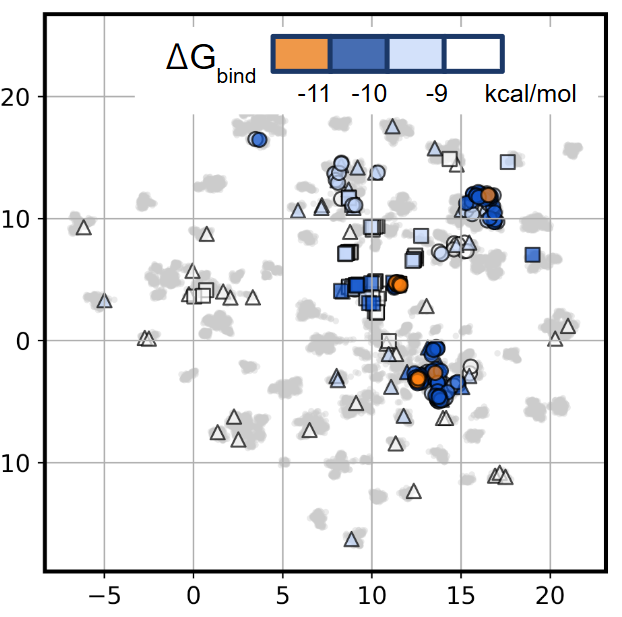
図 4. AL-FEPの各サイクルで選択・評価した化合物の化合物空間プロット。化合物空間は化合物プール内の全化合物のMorgan FingerprintをUMAPにより次元削減して定義した。マーカー△、□、〇は、それぞれRandom、Exploration、Exploitation戦略で選択された化合物を示す。
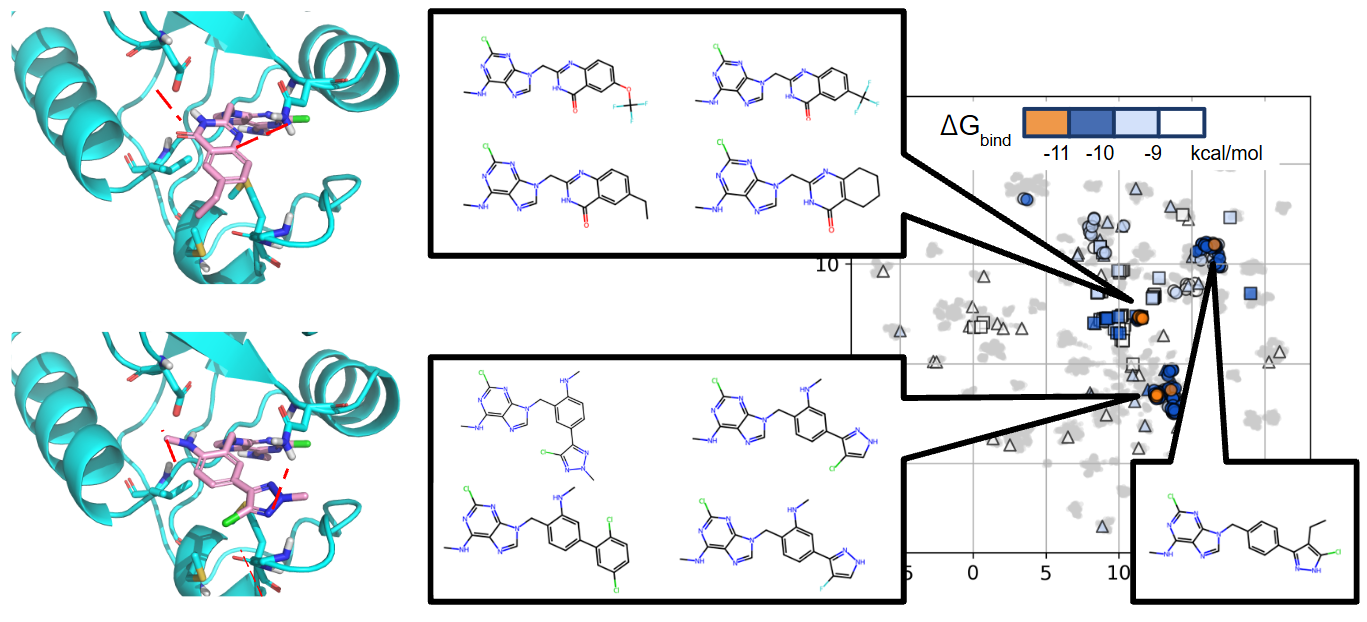
図 5. 結合が強い化合物の構造例。
2. 探索効率の比較
次に、AL-FEP戦略で選択した360化合物の\(\Delta G_{\text{bind}}\)分布を、化合物プールからランダムに360化合物を選択した場合と比較しました(図6)。文献で報告されている最も阻害活性の強い化合物の\(\Delta G_{\text{bind}}\)は -9.6 kcal/mol (IC50=0.18μM)です。AL-FEPは、この既知の強力な化合物を上回る (\(\Delta G_{\text{bind}}\) < -10 kcal/mol )有望な候補を、ランダム選択に比べて圧倒的に効率良く見出せることが示されました。
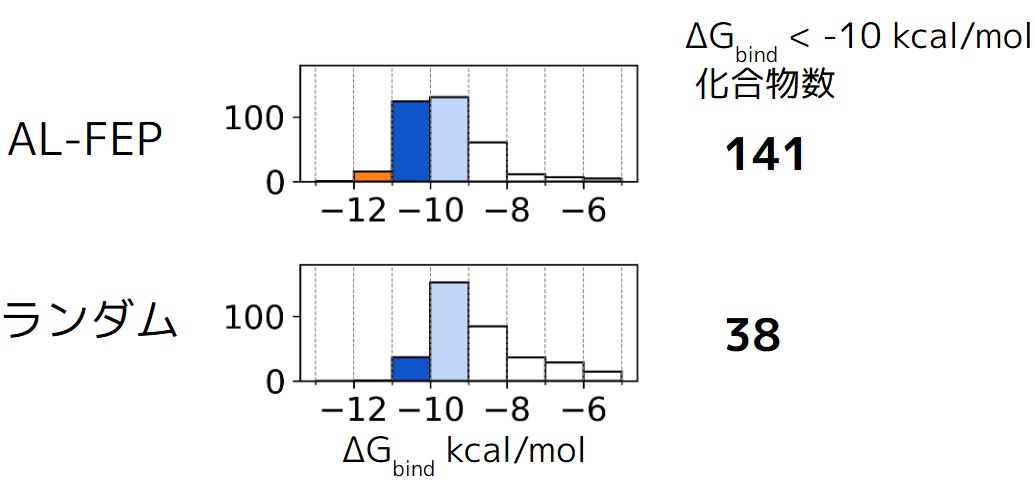
図 6. AL-FEP (360化合物 )とランダム選択 (360化合物 )の\(\Delta G_{\text{bind}}\)分布の比較。
まとめと展望
AL-FEPワークフローは、Active Learning (AL) による効率的な化合物空間探索とRBFEPによる高精度な結合自由エネルギー計算を組み合わせ、「Computational DMTA cycle」を構築します。本事例では、強い活性が期待できるYTHDC1阻害剤の候補を、全化合物の約2%という限られた計算リソース内で効率的に特定できました。これは、リード最適化における本手法の有効性を示していると言えます。
今後は、さらなるP-FEPの機能拡張を推進し、創薬の化合物デザインプロセスを高度に自動化することで、創薬プロセス全体の生産性向上を目指します。
References
[1] P-FEP (RBFEP計算サービス) 提供開始, Preferred Networks社ブログ https://tech.preferred.jp/ja/blog/pfep-launch/
[2] Wang, L., et al. (2015). “Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field.” J Am Chem Soc 137(7): 2695-2703.
[3] Thompson, J., et al. (2022). “Optimizing active learning for free energy calculations.” Artificial Intelligence in the Life Sciences 2: 100050.
[4] Gorantla, R., et al. (2024). “Benchmarking Active Learning Protocols for Ligand-Binding Affinity Prediction.” J Chem Inf Model 64(6): 1955-1965.
[5] Zalesak, F., et al. (2024). “Structure-Based Design of a Potent and Selective YTHDC1 Ligand.” Journal of Medicinal Chemistry 67(11): 9516-9535.
[6] Fialkova, V., et al. (2022). “LibINVENT: Reaction-based Generative Scaffold Decoration for in Silico Library Design.” J Chem Inf Model 62(9): 2046-2063.
—
本記事およびP-FEPに関するお問い合わせ先:drug-info[a]preferred.jp
[a]を @ に置き換えてご送信ください。
Tag