Blog
本記事は、2022年夏季インターンシッププログラムで勤務された古井海里さんによる寄稿です。
PFN2022年夏季インターンに参加した東京工業大学修士1年の古井海里です。大学ではバイオインフォマティクス・ケモインフォマティクスに関する研究をしています。本インターンでは「創薬に関する機械学習や分子シミュレーションの応用研究」のテーマで構造ベースのバーチャルスクリーニングのための機械学習スコアリング関数の汎化性能の評価を試みました。
背景
創薬では、1つの薬を上市するまでに数十億ドルもの開発費用や、十年程度の開発期間要すると言われており、そのコストは年々増加しています[1]。その主な理由として低分子が扱いやすいシンプルな創薬ターゲットの枯渇が挙げられます。また、薬剤になりうる化合物の数は10^20~10^60程度存在すると言われており[2]、広範な化合物空間から標的との活性がある化合物を見つけるのは非常に難しいことです。
創薬の初期段階では、薬剤候補化合物を探索するためにスクリーニング実験を行い、ヒット化合物の同定を行います。大規模な化合物ライブラリから計算機を用いてアッセイで活性を持ちうる化合物を絞り込むことをバーチャルスクリーニングと呼びます。バーチャルスクリーニングの精度向上は、膨大なアッセイの実施に伴うコストを削減するために重要です。
バーチャルスクリーニングは主に、標的との既知の活性情報に基づき、機械学習などによって活性予測をするリガンドベースの手法と、標的タンパク質の立体構造情報を用い、ドッキングや分子シミュレーションを行う構造ベースの手法に大別されます。構造ベースによる手法は、既知の活性情報を用いずにスクリーニングを行うことができるという利点がありますが、その予測精度は十分とは言い難く、その精度向上が盛んに試みられています。
その中の一つである機械学習ベースのスコアリング関数(Machine learning-based scoring funcion; MLSF)は、既知の結合親和性情報を学習した機械学習モデルによって結合ポーズを評価します[3]。このMLSFに対して、ランダムフォレスト(RF)、勾配ブースティング決定木、深層学習(畳み込みニューラルネットワーク、グラフニューラルネットワーク等)の様々な機械学習手法が適用されています[3]が、特に深層学習ベースのMLSFがスクリーニングタスクや結合能予測など様々なベンチマークにおいて、既存手法よりも高い性能を達成すると報告されています[4]。
MLSFの汎化性能評価
MLSFの予測精度は高いとされている一方で、ZhuらはこれまでのベンチマークがMLSFを楽観的に評価している可能性を指摘しています[5]。
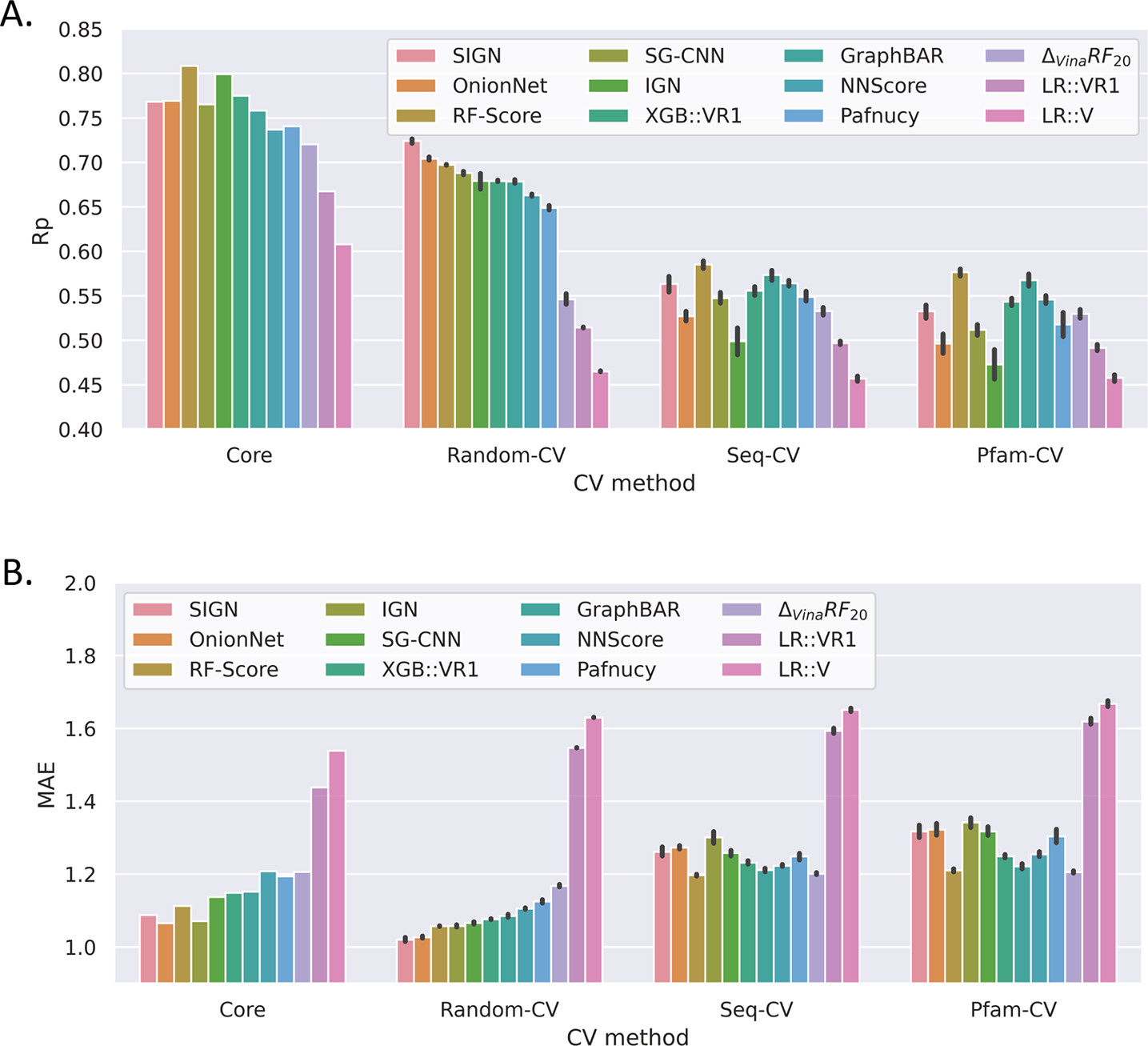
図1 MLSFに対するデータ分割ごとの交差検証([5]より引用)
Zhuらは、PDBBindデータセットを用いた結合親和性予測タスクに関するMLSFの汎化性能を評価しました(図1)。結果として、ランダム分割に基づく交差検証(Random-CV)ではSIGN[12]やOninonnet[13]などの深層学習ベースのスコアリング関数が高い予測精度を示しているのに対して、配列類似度(Seq-CV)やドメイン(Pfam-CV)に基づいてクラスタリングし交差検証を行った場合には、RF-Score[10]と呼ばれるランダムフォレストベースのMLSFが最も良いことが示されています。
Random-CVでのみ高い精度を示すMLSFは、訓練セットに含まれる類縁タンパク質の情報のみ、あるいは類縁タンパク質の活性化合物に共通する構造のみに基づいて検証データの結合能予測ができてしまうモデルであるということを意味しています。このようなMLSFが一般的なタンパク質−リガンド相互作用を捉えているとは言えません。
機械学習では、テストデータと訓練データの分布が同じであるという仮定が重要ですが、MLSFの一般的なユースケースにおいてはそれが困難な場合がしばしばあります。
したがって、MLSFを活用するためには汎用的にタンパク質-リガンド相互作用を捉えることが不可欠であり、その評価のためのデータ分割は適切に行うべきであるといえます。
研究目的
バーチャルスクリーニングでは可能性のある化合物データセットの中から正しく活性化合物を絞り込む、スクリーニング性能が重要です。Zhuらの検証がこのスクリーニングタスクにおいても有用かどうかを検証すべきだと考えました。
そこで本研究では、DUD-Eセット[6]と呼ばれる化合物スクリーニングのための一般的なベンチマークセットを用い、デコイ識別タスクに関する汎化性能の評価を試みました。加えて、DUD-Eセットには既知のアナログバイアスが知られているため[7]、MLSFの適切なベンチマークのためにTocoDecoy[8]と呼ばれるデコイ生成方法による評価も同時に行いました。
実験方法
データセット
DUD-Eセット
DUD-Eセットは、102のタンパク質標的に関する既知の活性化合物と、活性化合物と物理化学特性が類似するトポロジーの異なる50のデコイに関するデータセットであり、構造ベースのバーチャルスクリーニング性能の一般的なベンチマークとして用いられています。
本実験ではDUD-Eセットについて、ランダム分割とサブセット分割の2つの分割法でMLSFの予測精度の評価を行いました。
ランダム分割
3つのサブセット全てについてタンパク質ごとにランダムに訓練/検証/テストを8:1:1の比率で分割したランダム分割の学習と比較しました。ただし、ランダム分割は前述したように、訓練セットのタンパク質やリガンド構造の類似性のみに基づいてある程度予測ができてしまうため、MLSFモデルの性能を過大評価してしまう可能性があります。
サブセット分割
サブセット分割では、訓練セットにテストセットのタンパク質と類似した標的が含まれないようにすることで、汎用的にタンパク質−リガンド間の相互作用を学習できているか正しく評価することを目的としています。DUD-Eデータセットに含まれるGPCR、 Kinase、 Proteaseの3つのサブセットごとに訓練・テストセットの分割を行い(表1)、そのスクリーニング性能を評価しました。
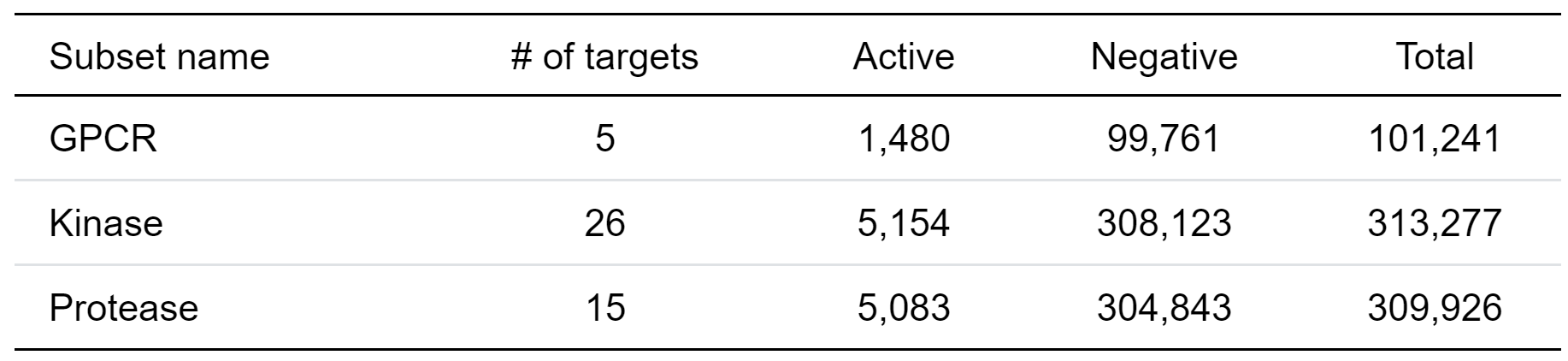
表1 DUD-Eセットにおける各サブセットの構成
このとき、訓練セットでは、Kinaseサブセットの検証セットを評価する際には、GPCRサブセットのみ訓練に用いる、Proteaseのみを訓練に用いる、GPCRとProtease両方のサブセットを訓練に用いる、の3ケースで評価を行います。
このように分割することで、例えば、テストセットには(訓練セットに含まれる)Kinaseサブセット内のタンパク質と類似したタンパク質や、Kinaseサブセットの標的に結合しやすいリガンドに共通する構造を含んでいないため、リガンドとその相互作用残基の特徴のみから学習できているかどうかを評価することができます。なお、サブセットは標的ごとに5分割して訓練・検証セットとしています。また、デコイ化合物はアクティブと同じ化合物数になるようにランダムに選択しました。
TocoDecoyセット
DUD-Eセットは構造ベースのベンチマークとして良く用いられていますが、すでに述べたように、隠れたアナログバイアスが存在していることが指摘されています[7]。図2は各DUD-Eサブセットの全てのデコイ同士のtanimoto類似度の比較をした結果ですが、活性化合物同士のtanimoto類似度の分布に対して、異なるサブセットに類似性の高いデコイが含まれていることが分かります。これは、機械学習モデルが訓練セットのデコイと構造的に似た化合物をタンパク−リガンド間の相互作用を見ずにデコイだと判別できてしまうということを意味します。
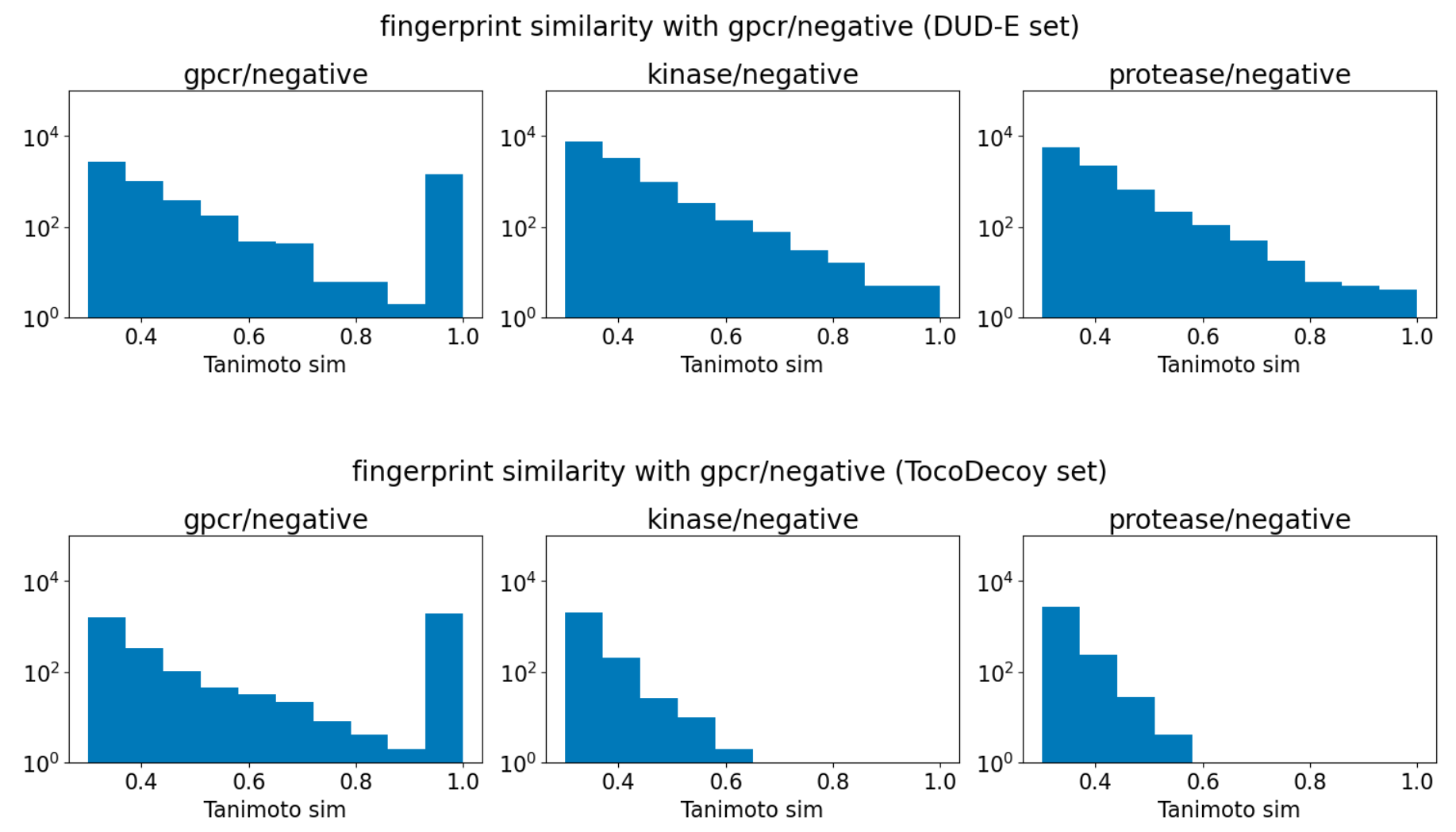
図2, 3 DUD-Eセット(上)とTocoDecoyセット(下)における 各デコイのGPCRのデコイとのtanimoto類似度。類似度計算のためのFingerprintとしてはECFP4-2048 bitを使用。
そこでDUD-Eセットの活性化合物と組み合わせてTocoDecoyのデコイ分子を活用する方法を考えました。TocoDecoyでは、条件付きRNNによる分子生成によって、活性化合物とトポロジーが異なるが類似した物理化学特性を持つ化合物を生成しています[8]。図3のように、TocoDecoyによって生成されたデコイ分子は異なるサブセットについて類似性が十分に低く、DUD-Eベンチマークで指摘されていたアナログバイアスを除去した評価をすることができると期待できます。ここでは、学習・検証についてはDUD-Eセットと同様にサブセットに基づく5分割の検証を行いました。
結合ポーズの生成
DUD-EやTocoDecoyデータセットでは、活性化合物のタンパク質に対する真の結合ポーズは用意されていないため、ドッキングによって結合ポーズの生成を行いました。結合ポーズの生成にはQuick Vina2[9]を用い、DUD-Eセット全ての活性化合物・デコイ化合物の結合ポーズを準備しました。ただし、同じタンパク質でも異なるリガンドに対する複数の構造が報告されているケースが多数あります。また、生成された活性化合物の結合ポーズはドッキングソフトウェアに依存してしまう可能性もあります。今回は、あくまでVinaによって得られたポーズを学習・評価しているということに留意する必要があります。
機械学習モデル
限られた期間で実験を行う都合上、PDBBind Coreセットで高い性能を示したRF-Score[10]とInteractionGraphNet (IGN) [4]の2つの異なるモデルについてのみ性能を評価しました。
RF-Score
RF-ScoreはランダムフォレストベースのMLSFです。特徴量にはAutoDock Vina[11]で用いられる6つの項と、タンパク質-リガンド間の原子の36の相互作用が用いられます。
InteractionGraphNet
InteractionGraphNet (IGN) はグラフベースのNNモデルに基づくMLSFです。タンパク質原子グラフ・リガンド原子グラフ・タンパク質−リガンド相互作用に関する原子グラフの3つのグラフを用いて相互作用を予測します。
実験結果
学習
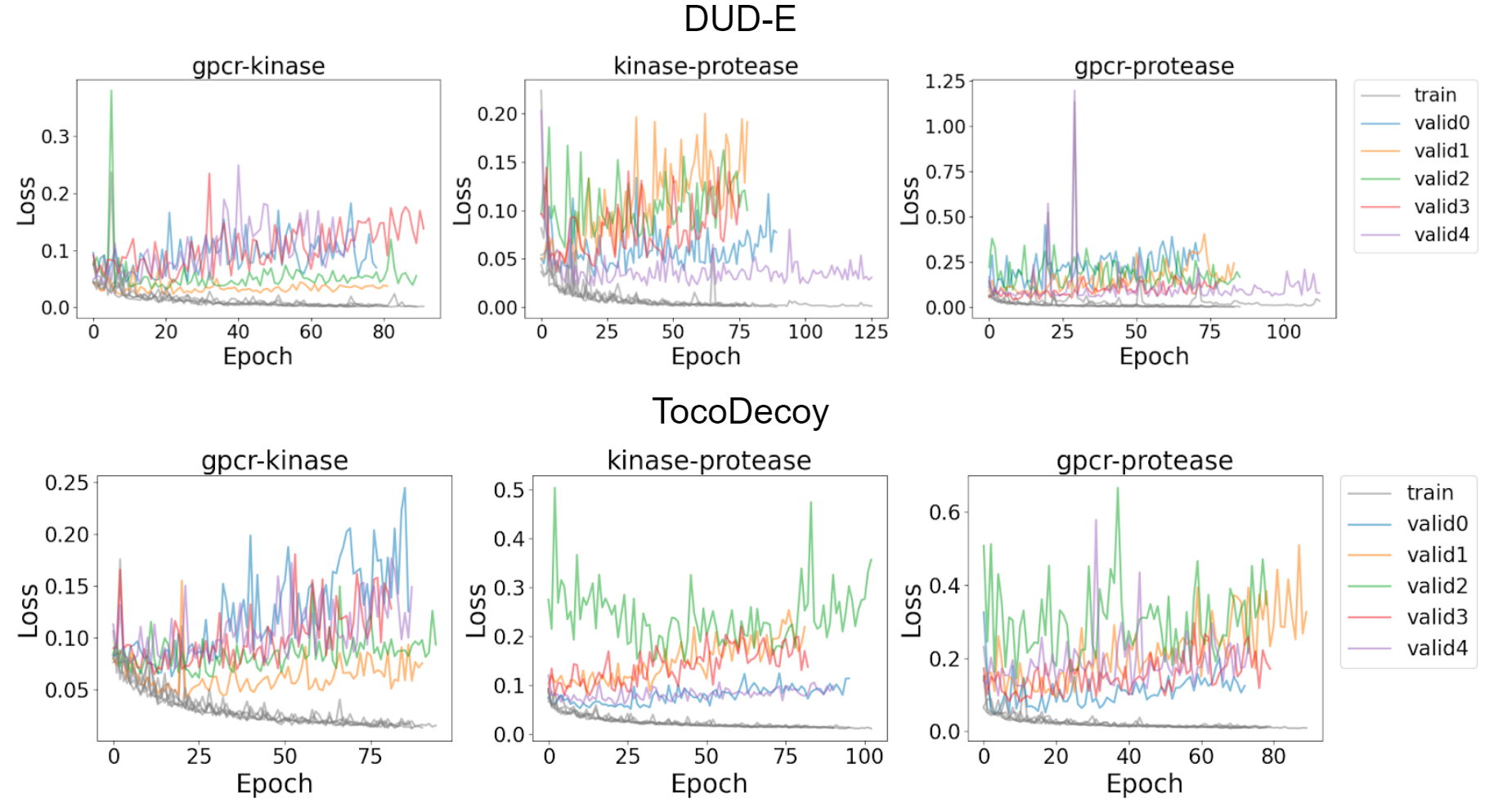
図4, 5 DUD-Eセット(上)とTocoDecoyセット(下)に対するIGNの学習曲線
図4、図5は複数のサブセットを用いてIGNを訓練したときの学習曲線を示しています。trainは各分割セットでの学習セットでの損失を示し(すべて同色)、valid0からvalid4は各分割の検証セットでの損失を示しており、70 epoch以上検証セットに対して予測が改善しないときに学習を終了させています。データセットによっては学習が進むにつれて検証セットの損失が大きくなっていることが分かります。
ランダムとサブセット分割時の評価比較
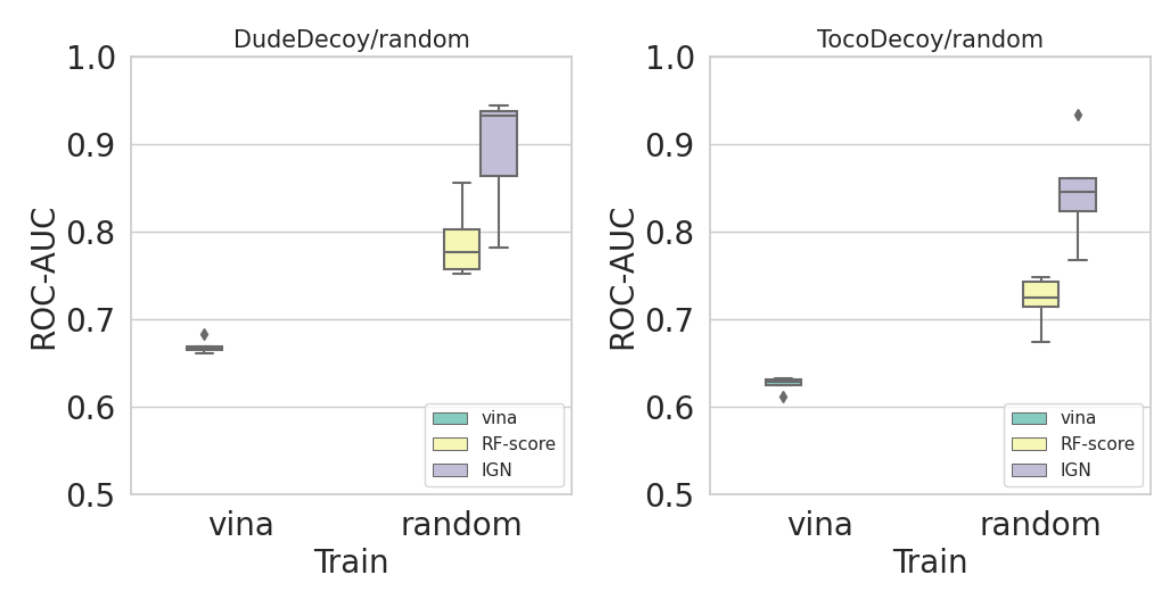
図6, 7 DUD-Eセット(左)TocoDecoyセット(右)に対するランダム分割時のROC-AUC値
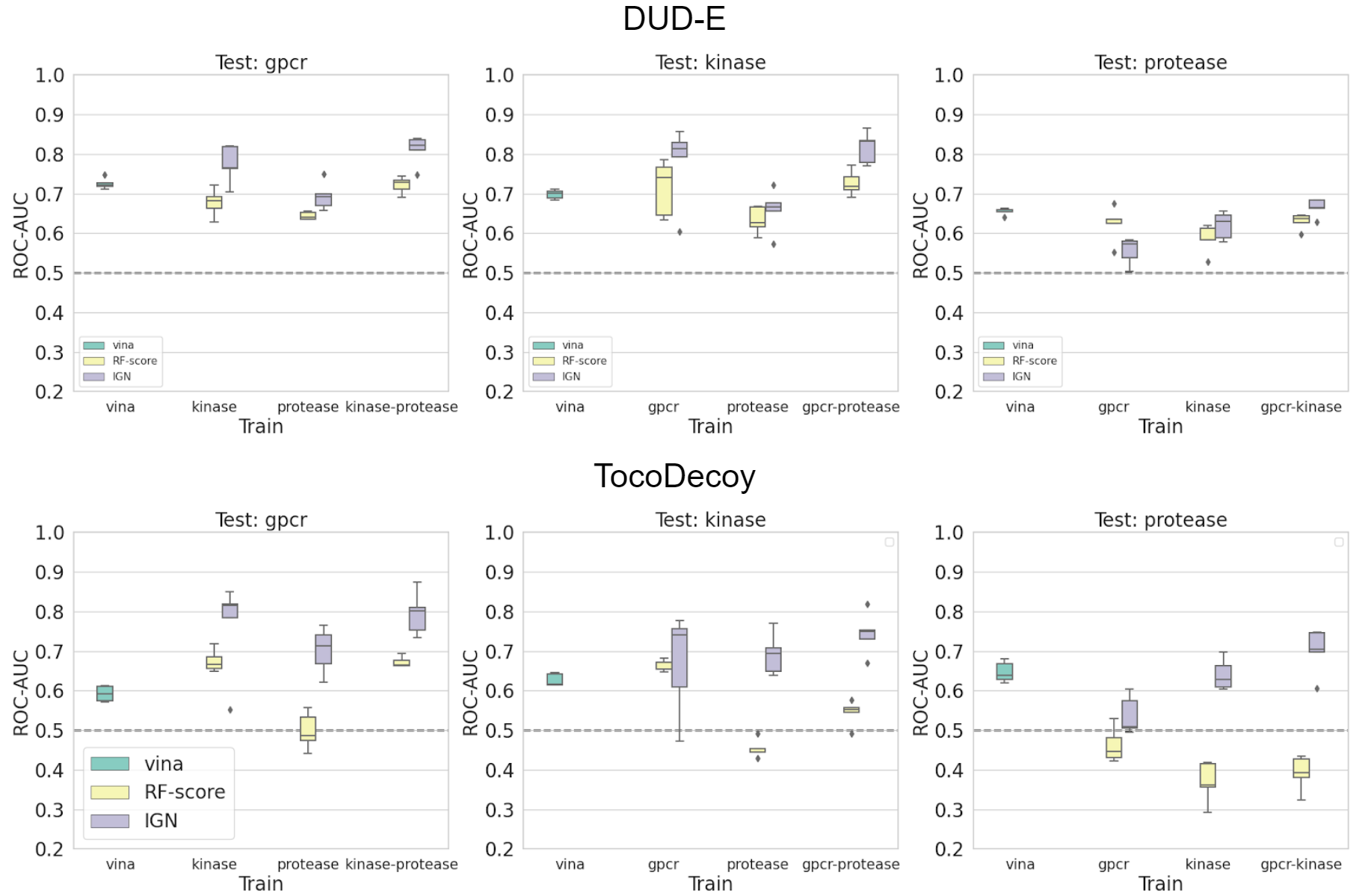
図8, 9 DUD-Eセット(上)とTocoDecoyセット(下)に対するサブセットに基づいた分割時のROC-AUC値
図6、図7より、ランダム分割のケースではIGNが特に高い予測性能を出していますが、図8、図9を見ると、サブセットベースで分割したときの予測精度はそこまで高くないことが分かりました。デコイ識別タスクにおいてもランダム分割が楽観的な予測をしてしまうため、適切なデータの分割が重要である事が示唆されます。なお、テストデータと同じサブセットを訓練データに用いた場合の予測精度はほとんど1に近い値になります。また、kinaseサブセットをテストセットとした場合を例に取ると、GPCRサブセットのみ、proteaseサブセットのみを訓練に用いるよりもGPCRとproteaseを組み合わせた場合の方が良い予測精度を示しており、汎化性能のためには幅広い標的を用いる事が有効であると示唆されました。なお、複数のサブセットを用いた場合、IGNの予測精度は単純にquick vina2が出力するVinaスコアを用いるケースよりも良い性能を示しました。TocoDecoyセットについてもDUD-Eセットとほぼ同様の結果が得られました。大きく異なる点として、kinase、proteaseサブセットをテストセットとしたときのRF-ScoreのROC-AUCが0.5を下回るケースがあり、訓練セットに過適合している可能性が示唆されました。全体として、Zhuらの結果[5]と異なりRF-ScoreよりもIGNの方が多くのケースで性能が良いという結果が得られました。
結論
本インターンでは、DUD-EベンチマークとTocoDecoyを用いてMLSFのデコイ識別タスクにおける汎化性能の評価を行いました。結果として、デコイ識別タスクにおいてもランダム分割による評価方法が楽観的な結果を与えてしまうことが示されました。一方で、サブセットに基づいたデータ分割や、TocoDecoyで生成したアナログバイアスのないデコイを用いた評価方法では、MLSFの汎化性能を適切に評価できている可能性が示されました。
おわりに
自分の都合で従来よりも短い期間でのインターンシップではありましたが、社員の方々の丁寧なサポートや交流の機会を頂いたおかげで非常に密度の高い経験ができました。また、メンターの石谷さん、副メンターの力丸さん、富田さんには、毎日のミーティングで様々なアドバイスを頂いたり、トラブル対応に協力していただいたおかげで何とかインターン期間中にテーマをまとめることができました。誠にありがとうございました。
参考文献
[1] J. A. DiMasi, et al., J. Health Econ. 47, 20–33, 2016. (doi: 10.1016/j.jhealeco.2016.01.012)
[2] P. G. Polishchuk, et al., J. Comput. Aided Mol. Des. 27, 675–679, 2013. (doi: 10.1007/s10822-013-9672-4)
[3] C. Yang, et al., Molecules 27, 4568, 2022. (doi: 10.3390/molecules27144568)
[4] D. Jiang, et al., J. Med. Chem. 64, 18209–18232, 2021. (doi: 10.1021/acs.jmedchem.1c01830)
[5] H. Zhu, et al., J. Chem. Inf. Model. 62, 5485–5502, 2022. (doi: 10.1021/acs.jcim.2c01149)
[6] M. Mysinger, et al., J. Med. Chem. 55, 6582–6594, 2012. (doi: 10.1021/jm300687e)
[7] J. Sieg, et al., J. Chem. Inf. Model. 59, 947–961, 2019. (doi: 10.1021/acs.jcim.8b00712)
[8] X. Zhang, et al., J. Med. Chem. 65, 7918–7932, 2022. (doi: 10.1021/acs.jmedchem.2c00460)
[9] A. Alhossary, et al., Bioinform. 31, 2214–2216, 2015. (doi: 10.1093/bioinformatics/btv082)
[10] H. Li, et al., Mol. Inform. 34, 115-26, 2015. (doi: 10.1002/minf.201400132)
[11] O. Trott, et al., J. Comput. Chem. 31, 455–461, 2010. (doi: 10.1002/jcc.21334)
[12] S. Li, et al., Proc. 27th ACM SIGKDD Int. Conf. Knowl. Discov. Data Min. 975– 985, 2021. (doi: 10.1145/3447548.3467311 )
[13] L. Zheng, et al. ACS Omega 4, 15956–15965, 2019. (doi: 10.1021/acsomega.9b01997)
Tag