Blog
本記事は、2023年夏季インターンシッププログラムで勤務された東一織さんによる寄稿です。
はじめに
2023年夏季インターンシップに参加した東京大学大学院薬学系研究科博士課程1年の東一織です。研究室では、臨床情報やオミクスデータに対する機械学習の研究を行っています。今回のインターンでは、「遺伝⼦に関するグラフを利⽤したモデルの開発 」に取り組みました。
背景
生物学におけるマルチモダリティデータ
遺伝情報が「DNA→(転写)→RNA→(翻訳)→タンパク質」の順に伝達・変換される一連の流れはセントラルドグマと呼ばれ、生命科学における重要な概念の一つです。
近年のシングルセル解析技術の進展に伴い、同じ細胞から複数のモダリティ(クロマチンアクセシビリティ (*1)、RNA、タンパク質など)を測定することが可能となり、細胞の転写や機能的プロセスについての理解が進んでいます [1] 。
一方で、シングルセル解析によるマルチモダリティデータの取得は費用が高く、技術的にも黎明期であることから、ATAC-Seq (*2) やRNA-Seq (*3) などの単一モダリティデータの取得が未だに主流です。そのため、入手可能な単一モダリティより別モダリティを予測する手法は、複雑な生体応答の背景にあるモダリティ間の関係性を理解するという点で重要です。
生物学的プロセスとグラフ構造
モダリティ間の関係性を予測する上で、生体を秩序立てている背景(生物学的プロセス)を理解する必要があります。特に重要な考え方が、遺伝子やタンパク質などのモダリティ構成要素は多くの手段によって制御されているというものです。
具体的には、転写制御ネットワークやタンパク質相互作用などが挙げられます。これらの生体分子グラフによる制御・調整過程は「生物学的プロセス」の一つであり、セントラルドグマをはじめとする複雑な生命現象を理解する上で有用な情報となります。
グラフ表現学習
さて、生体分子グラフを生かしてどのようなモデルを構築するのが良いでしょうか?Graph Neural Network(GNN)は代表的な深層学習パラダイムであり、グラフの表現学習のための効果的なフレームワークです [2]。GNNでは通常、隣接するノードの情報をもとに各ノードの特徴量を更新し、最終的に、全てのノードの特徴表現を集約することでグラフ全体の特徴表現を生成します。
今回は、GNNを実際の細胞に由来する生物データへと適用を試みます。そこで、現実世界のデータは、複数の種類のノードとエッジに由来するHeterogeneous Graph (ヘテログラフ)として捉える考え方に着目しました。例えば、レコメンデーション分野では顧客と商品は異なるノードで表現され、ソーシャルネットワークでは個人間のフォローやブロックなどの異なる関係性を有します。
このように、ヘテログラフの特性を生かしたGNNは、包括的な情報を活用できると期待され近年研究が進んでいます。一方で、生物学におけるモダリティ間の関係性予測への適用は限定的であり、特に、少数サンプルで学習した際の頑健性や、どのようなグラフを考慮するべきかについての言及はほとんどありませんでした。
そこで今回のインターンシップでは、遺伝子に関するグラフ構造を含む、細胞を取り巻くヘテロな関係性を反映するGNNを構築し、その効果の検証を行いました。
手法
実装のベース
実装のベースの手法として2022年に提案されたScMoGCN [3] というモデルを採用しました。モデル構造は図1に示す通りです。本モデルでは、与えられたモダリティよりCell-Featureグラフを構築し、同時にFeature-Featureグラフを外部情報として組み込んだヘテログラフを想定しています。ここで、例えばRNAモダリティの入力を想定する場合では、Cell-Featureグラフのエッジの重みは遺伝子の発現量を、Feature-Featureグラフについては遺伝子間の相互作用を意味します。このように2種類のグラフを区別して畳み込んで細胞ノードの特徴量を更新することで、別モダリティ(タンパク質発現量)を精度よく予測できることが報告されています。
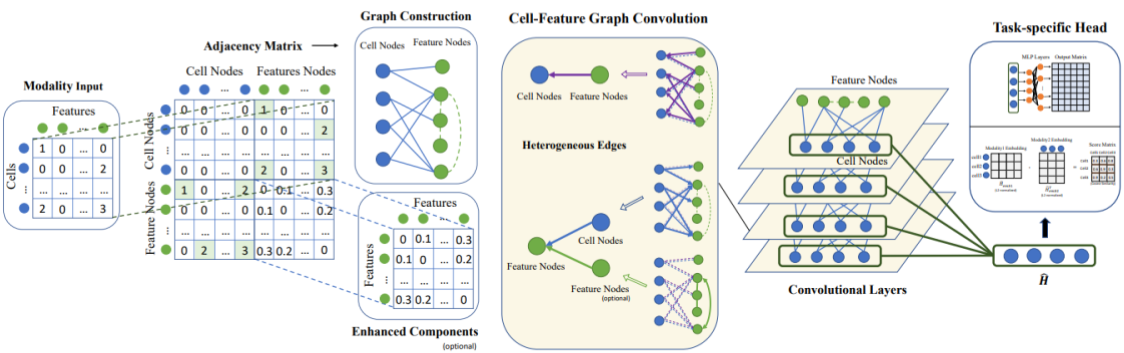
図1: ScMoGCNのモデル構造(文献 [3] Fig.1より抜粋。一部改変。)
Feature-Featureサブグラフを区別して畳み込む
さて、先のモデルでは、Feature-Featureグラフを一つの大きなグラフとして捉えていました。一方で、特定の生物学的プロセスを担う遺伝子は限られており、同一遺伝子であってもその役回りは異なると考えられます。そのため、Feature-Featureグラフ自体も、生物学的プロセスと対応するいくつかのサブグラフに由来するヘテログラフとして捉え、ノードの特徴量を区別して畳み込む以下の実装を追加しました(図2)。
\[ h_{i}^{l+1} = h_{i}^{l} + \alpha \cdot \left( \sum_{k=1}^{K} m_{V_{k} \rightarrow V_{k}}^{i, l} \right) + (1 – \alpha) \cdot m_{U \rightarrow V}^{i, l} \]
ここではfeature nodeの特徴量の更新を示しており、\( h_{i}^{l} \) はl層目のi番目のnodeの特徴量、\( m_{V_{k} \rightarrow V_{k}}^{i, l} \)はk番目のFeature-Featureサブグラフにおけるメッセージパッシング、また、\( m_{U \rightarrow V}^{i, l} \)はCell-Featureグラフにおけるメッセージパッシングを表します。このように生物学的プロセスを区別してFeatrureノードを更新することで、より解釈性の高い特徴量を確保できると期待されます。
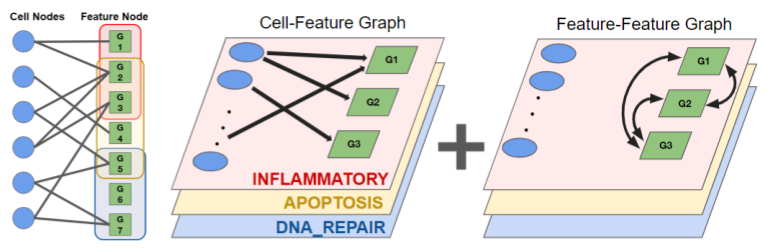
図2: Featureノードの特徴量の更新。ここではINFLAMMATORY, APOPTOSIS, およびDNA_REPAIRの3つの生物学的プロセスに由来するグラフを区別して畳み込んでいる。
データセット
ベンチマークデータとして、同一細胞に異なるモダリティ情報が付帯したデータが必要となります。今回のインターンでは、Kaggleの「Open Problems – Multimodal Single-Cell Integration」[4] 内のデータを選択しました。本データセットには、RNAと膜タンパク質発現量を同一細胞から測定したCITE-Seqの測定値と、DNA(クロマチンアクセシビリティ)とRNAを同一細胞から測定したChromium Single Cell Multiome ATAC + Gene Expression (Multiome) の測定値が含まれます。
Kaggleのコンペと同様に、① DNAからRNAの予測と、② RNAからProteinの予測の2つの異なるモダリティ間の関係性を予測するタスクに取り組みました(図3)。なお、均質なコホートでの解析を実施するため、特定の被験者における特定の日時に取得した細胞を解析対象としました。コンペに習い、性能評価指標には相関係数を採用しましたが、特定の特徴量を予測した際の細胞ワイドな相関係数で評価するなど、コンペ内での評価指標と必ずしも一致しない点に留意してください。
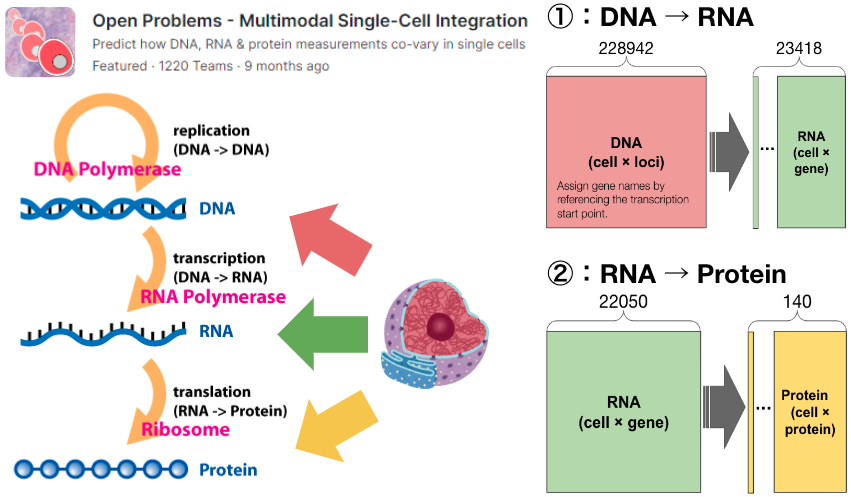
図3: 異なるモダリティ間の関係性予測
結果・考察
RNA → Protein の予測
まず初めに、CITE-Seqによる測定データを用いて、RNAの発現量から特定の膜タンパク質発現量を予測するタスクに取り組みました。複数の細胞種に渡って比較的高い発現量を示し、その生理学的機能もよく研究されているCD47やCD44などを予測対象のタンパク質として例に挙げています。
モデルに組み込むFeature-Featureグラフにはいくつか定義の仕方がありますが、ここでは、Molecular Signature Database (MsigDB) [5] よりHallmarks遺伝子セットを取得し、学習データにおける遺伝子同士の発現量の相関係数をエッジの重みとして有するグラフを構築しました。このグラフは、生物学的プロセスとその相互作用情報を有することから、ある種のpathway (*4)と見なすことが可能でしょう。
そこで、CD47発現量制御に重要とされるIFN-αおよびIFN-γ、また、CD44を応答因子として含むエストロゲン応答を具体的なpathwayとして選択し、グラフとしてモデルに組み込んだ際の有用性の評価に取り組みました。
その結果、学習サンプルが十分なケースでは、グラフ構造の考慮の有無に寄らず推定性能に大きな改善は見られず、baselineであるLightGBMとも同等でした(表1)。
表1: 全ての訓練データを用いて学習した際の推定性能 (相関係数で評価)
| n=7609 | CD47 | CD44 | ||||||
| w. pathway | w/o. pathway | LightGBM | w. pathway | w/o. pathway | LightGBM | |||
| IFN-α | 0.536 | 0.525 | 0.556 | 0.493 | 0.494 | 0.523 | ||
| IFN-γ | 0.533 | 0.526 | 0.532 | 0.542 | 0.545 | 0.533 | ||
| Estrogen Res Early | 0.511 | 0.499 | 0.523 | 0.529 | 0.541 | 0.531 | ||
| Estrogen Res Late | 0.532 | 0.527 | 0.537 | 0.570 | 0.567 | 0.575 | ||
一方で、ランダムに100サンプルを選択した少数のデータで学習した際には、特定のグラフ構造を用いることで性能の改善が確認され、より頑健な予測を達成しました(図4および表2)。具体的には、IFN-αやIFN-γなど炎症応答関連pathwayを用いた際には、CD47タンパク質の予測性能が特異的に改善し、エストロゲンに対する応答関連pathwayを用いた際にはCD44のみが予測性能の改善を示しました。
ただ単にグラフ構造を組み込むことで性能が向上するのではなく、予測対象の発現量制御との関与が知られるpathwayが予測対象特異的に性能向上に寄与した点は興味深いです。
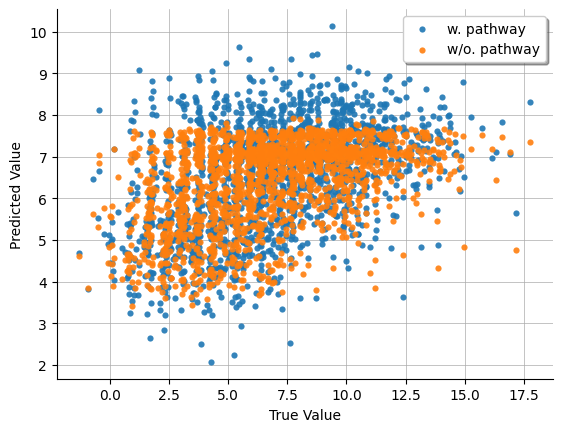
図4: 少数サンプルを用いて学習した際の、IFN-α関連pathwayの考慮の有無によるCD47タンパク質の予測値と実測値
表2: 少数の訓練データを用いて学習した際の推定性能 (相関係数で評価; 複数回試行した際の平均値と標準偏差)
| n=100 | CD47 | CD44 | ||||||
| w. pathway | w/o. pathway | LightGBM | w. pathway | w/o. pathway | LightGBM | |||
| IFN-α | 0.358±0.028 | 0.308±0.087 | 0.309±0.003 | 0.314±0.082 | 0.318±0.105 | 0.279±0.005 | ||
| IFN-γ | 0.385±0.032 | 0.351±0.045 | 0.296±0.005 | 0.334±0.123 | 0.377±0.025 | 0.312±0.004 | ||
| Estrogen Res Early | 0.307±0.092 | 0.320±0.109 | 0.257±0.004 | 0.317±0.051 | 0.268±0.123 | 0.266±0.002 | ||
| Estrogen Res Late | 0.353±0.060 | 0.358±0.045 | 0.293±0.004 | 0.323±0.089 | 0.301±0.135 | 0.287±0.007 | ||
次に、IFN-α、 IFN-γ、 およびInflammatory responseの3つの炎症に関連するpathwayを連続して畳み込むモデルを、少数サンプルで学習した際の性能を比較しました。
表3: 炎症に関連する3つのpathwayを連続して畳み込んだ際の予測性能 (相関係数で評価; 複数回試行した際の平均値と標準偏差)
| n=100 | w. pathway | w/o. pathway | LightGBM | |
| CD47 | 0.428±0.033 | 0.408±0.045 | 0.310±0.004 | |
| CD38 | 0.477±0.054 | 0.483±0.051 | 0.510±0.004 | |
| CD45 | 0.333±0.046 | 0.325±0.033 | 0.233±0.003 |
炎症応答に関連するCD47タンパク質の発現量の予測については、僅かに評価値は向上したものの、期待していたほどの大きな改善ではありませんでした。また、CD38やCD45など、免疫応答に関与する他のタンパク質の予測についても改善への寄与は限定的でした(表3)。この要因として、複数のpathwayを考慮した場合には含まれる遺伝子数も増加するため、グラフを用いずとも回帰するのに十分な安定した特徴量が取得できていたと推察されます。
さて、以上の結果は、遺伝子群の事前情報より解析対象の発現量の相関を重みとするソフトなグラフ構造を構築するケースでした。次に、モデルに使用するFeature-Featureグラフを明示的なグラフ構造として入力した場合についても同様の評価を行いました。ここでは、予測するタンパク質と同名の遺伝子に着目し、GENEMANIA [6] より当該遺伝子を中心とする相互作用情報をグラフとして取得しました(図5)。本ケースでも同様に、学習に使用するサンプル数が少ない際には、グラフ構造を反映したモデルを用いることでより頑健な予測を達成しました(表4)。
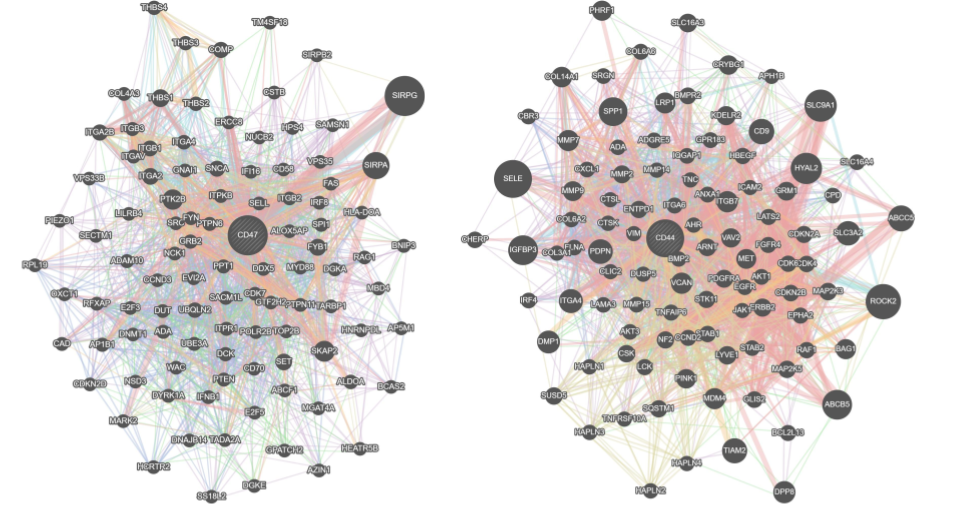
図5: GENEMANIA由来のグラフ構造。
表4: GENEMANIA由来のグラフ構造を用いた際の予測性能 (相関係数で評価; 複数回試行した際の平均値と標準偏差)
| protein | CD47 | CD44 | ||||||
| w. graph | w/o. graph | LightGBM | w. graph | w/o. graph | LightGBM | |||
| n=7609 | 0.462 | 0.454 | 0.450 | 0.521 | 0.509 | 0.548 | ||
| n=100 | 0.239±0.050 | 0.195±0.108 | 0.136±0.006 | 0.333±0.083 | 0.271±0.124 | 0.346±0.004 | ||
これまでの結果を総合すると、予測対象の制御に関与する生物学的プロセスをグラフ構造として考慮したモデルでは「少数サンプルかつ少数特徴量」の条件下で、より頑健な推定が可能であると結論付けられます。
比較的限られた条件ではありますが、臨床検査値などの検体数が少なく遺伝子の測定値も網羅的でないデータへの応用が期待されます。また、ランドマーク遺伝子と呼ばれる少数の遺伝子発現量プロファイルを格納したデータベースも整備されていることから、これらのデータ解析にも有効かもしれません。
DNA → RNA の予測
次に、Multiomeによる測定データを用いて、DNA(クロマチンアクセシビリティ)からRNAの発現量の予測についても同様に取り組みました。ここでは、入力するDNAのモダリティについて、転写開始点を参照することで遺伝子座位から遺伝子名へと変換し、遺伝子グラフ構造を考慮可能としました。
しかし、RNAからProteinを予測したタスクと異なり、本タスクでは少数サンプルかつ少数特徴量の条件下においても、グラフを考慮する有用性は認められませんでした。この結果については以下のように考察しています。
- 入力するDNAデータのスパース性
今回扱ったDNAのデータは非ゼロ要素が3%程度とスパースな性質を有し、Cell-Featureグラフの接続が局所的な構造になります。そのため、Featureノードの特徴量を遺伝子グラフなどを工夫して更新しても、最終的にCellノードに伝達される情報が限定され、サンプルを特徴づけられなかった可能性が考えられます。
- 予測対象のRNAにおける測定値の信頼性の問題
予測値(縦軸)と実測値(横軸)の関係性をプロットすると、多くのサンプルで予測対象遺伝子が発現していないことが分かります(図6)。しかし、これらの実測値のクラスターは、DNA側の値で層別化できなかったことから、シングルセルの測定の問題であると推察されます。Kaggle内の総括においても、各RNA予測における相関は中央値が0.075であったと言及されており [8] 、データの信頼性も含めて難しい問題設定であったと考えられます。
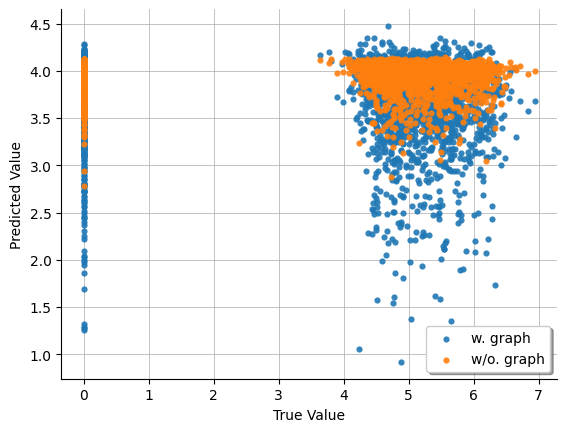
図6: 少数サンプルを用いて学習した際の、IFN-α関連pathwayの考慮の有無によるCD47タンパク質の予測値と実測値
まとめ・展望
今回のインターンでは、生物学的プロセスを反映する遺伝子グラフを個別に畳み込むヘテログラフニューラルネットワークを実装し、シングルセル由来データにおけるモダリティ間の関係性の予測性能を評価しました。RNAからProteinを予測するタスクにおいては、学習サンプル数が限られ、かつ使用できる特徴量が少ない状況においては、予測対象モダリティの制御に関与する特定のグラフ構造を用いることで性能の改善が認められました。一方で、サンプル数や特徴量が十分なケースでは本モデルを用いることで飛躍的な予測性能の改善は認められませんでした。
モデルのさらなる改善案としては、Cell-Featureグラフについて、Featureノードの特徴量の更新に応じてエッジの切り貼りを行うことなどが考えられます。本案は入力データの欠損値補完に近いものであり、細胞を特徴づける情報の流路が確保できると期待されます。
また、今回扱ったモデルでは、生物学的プロセスを「区別」はしたものの「同等」の重みで畳み込んでいます。一方で、Gene Ontology [7] に代表されるように、生物学プロセスには包含や階層の概念もあるため、類似度に応じた重みの変化などを組み込むことも予測性能の改善に寄与するかもしれません。
最後に、本インターンを通じて、シングルセルデータの取り扱いの難しさを痛感しました。スパースなデータの性質に加え、被験者や細胞種ごとの分布の違いなどの様々な交絡因子を考慮する必要がありました。そのため、意図しない結果が得られた際に、モデル側の問題なのか、あるいは問題設定を見直すべきかの判断が難しく、データに翻弄された印象があります。特に、細胞ごとに対応の取れたマルチモーダルなデータは入手可能なものが少ないため、今後の解析技術の向上と公開データの普及が望まれます。
終わりに
PFNでは幅広い分野で活発に研究活動が行われており、様々な分野の最新の知見に触れることができたのは貴重な体験でした。また、異分野で活躍する他のインターン生や社員の方との多角的な議論を通じ、研究への取り組み方について多くを学ぶことができました。
また、日々の取り組みでは、メンターの井形さん、副メンターの小寺さん、並びにオミクスチームの皆さんには、手厚いサポートをいただき、大変心強かったです。
約1ヵ月半という短い期間でしたが、充実したインターンシップとなりました。最後になりますが、この場を借りて御礼申し上げます。
参考文献
- Tang, Xin, et al. “Explainable multi-task learning for multi-modality biological data analysis.” Nature Communications 14.1 (2023): 2546.
- Xu, Keyulu, et al. “How powerful are graph neural networks?.” arXiv preprint arXiv:1810.00826 (2018).
- Wen, Hongzhi, et al. “Graph neural networks for multimodal single-cell data integration.” Proceedings of the 28th ACM SIGKDD conference on knowledge discovery and data mining. 2022.
- Daniel Burkhardt, Malte Luecken, Andrew Benz, Peter Holderrieth, Jonathan Bloom, Christopher Lance, Ashley Chow, Ryan Holbrook. (2022). Open Problems – Multimodal Single-Cell Integration. Kaggle. https://kaggle.com/competitions/open-problems-multimodal
- Subramanian, Aravind, et al. “Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles.” Proceedings of the National Academy of Sciences 102.43 (2005): 15545-15550.
- Franz, Max, et al. “GeneMANIA update 2018.” Nucleic acids research 46.W1 (2018): W60-W64.
- Ashburner, Michael, et al. “Gene ontology: tool for the unification of biology.” Nature genetics 25.1 (2000): 25-29.
- https://www.kaggle.com/competitions/open-problems-multimodal/discussion/372609
注釈
*1. クロマチンアクセシビリティ: DNAとヒストンの複合体であるクロマチンは、その凝集度合いが動的に変化することで転写制御を行う。特に、凝集が緩まった領域では分子のアクセス性が増すことで転写が活性化される。
*2. ATAC-Seq: クロマチンが緩まり露出したDNAを定量することで、転写が活性化されている部位を特定可能である。
*3. RNA-Seq: mRNAなどの転写物の配列情報を網羅的に読み取り、遺伝子発現を定量可能である。
*4. pathway: 生体内で秩序だった制御関係を示す一連の経路。遺伝子やタンパク質の相互作用をはじめ、シグナル伝達経路や代謝経路など幅広く用いられる。
Tag