Blog
2026.04.17
機械学習ポテンシャルの近似の限界を超えて【インターン募集】
So Takamoto
Head of Research Department, Materials & Drug Discovery Division
2026年もインターンの季節がやってきました。本稿は、機械学習ポテンシャルの研究分野の紹介を兼ねたインターン募集案内です。機械学習ポテンシャルはあくまでインターンに関連した研究テーマの一面ですが、具体例を通して私たちが普段どういった視点でテーマ設定を行っているのかの参考になれば幸いです。記事の内容は執筆者の高本が講演で発表した内容から一部を抜粋、加筆したものとなっています。
汎用機械学習ポテンシャル(Universal Machine Learning Interatomic Potential, Universal MLIP)の技術は近年大きく進展し、電子状態計算の限界を大幅に超えるスケールの分子動力学計算を活用して実用的な問題へ適用される事例も増えてきました。私たちは2019年から世界に先駆けてUniversal MLIPとしてPFPの開発をスタートし、2021年には汎用原子レベルシミュレータ「Matlantis」のコア技術として提供を開始しました。現在では多くのお客様にご活用いただいています。
MLIPの発展を支えてきた要因としては、他の機械学習分野と同様に、大規模データセットを構築可能にした計算資源の拡充や、複雑な関数近似を可能にしたニューラルネットワークの諸技術の進歩が挙げられます。
しかし本稿では、一旦それらの機械学習的な側面とは別の観点として、推論対象の物理量が機械学習によって近似可能なのか、近似可能であればどういったモデルであればよいのか、という点に着目してみたいと思います。
計算量のギャップ
近年の機械学習の発展は目覚ましく、あたかも任意の複雑な問題が学習可能であり、複雑な計算を回避できるかのような印象を与えることがあります。一方、機械学習モデルもアルゴリズムの一種である以上は理論的な限界があります。たとえば、線形時間で任意の巡回セールスマン問題の最適解を求める機械学習モデルを作ることは計算複雑性の観点から困難でしょう。こういった問題では、実用的な問題が持つ追加の制約をみて、解ける範囲への切り出しを行うことが機械学習モデルを設計する上で重要となってきます。
MLIPにも同様の議論が存在します。MLIPが通常ターゲットとするDFT(密度汎関数理論)計算は、電子数Nに対して計算量がおおむねO(N^3)に比例すると言われています。ここで計算量が3乗になるのは、電子が十分に緩和した状態を求めようとしているためです。
一方でMLIP、特にUniversal MLIPでは線形スケーリングO(N) を志向した設計が広く使われており、数値実験的に有効であることが示されています。この事実は、MLIPが学習対象としているエネルギーは電子状態計算のすべての情報を使わなくても良い精度で計算できるという都合の良い性質を持っていることを示唆しています。
このことは物理量の局所性の観点から考えることができます。理論的にはKohnのnearsightedness (density matrixが局在化し、遠方からの影響を受けにくい) と関係していますが、エネルギーは局所的な情報から求められる部分エネルギーの和としてよい精度で記述できる、ということが知られています。
経験的にもこの事実は広く受け入れられており、たとえば有機分子系では古典ポテンシャルは近接N体(しばしば4体まで)の相互作用としてしばしばモデル化されます。金属の場合には電子状態としては空間的に広がりうるのですが、それでもエネルギーという量に関しては経験的には局所近似は高精度に成立することが知られており、EAMポテンシャルなどの古典ポテンシャルによって有効性が検証されてきました。典型的にはEAMポテンシャルのカットオフ距離は0.5-0.6 nm程度となることが多いようです。
先述のようにMLIPは計算量の観点ではDFT計算を内部的に完全にエミュレートする能力を持っておらず、これがMLIPのスケーラビリティの源泉となっています。ある意味ではO(N) DFTの背景となるアイデアを援用しているともいえますが、それでもMLIPがしばしば「電子状態計算をバイパスしている」と表現されるのはこの関係に由来しています。グラフNNで多段階の相互作用を表現しているとはいえ、MLIPの相互作用1回あたりのカットオフ距離はしばしば1 nm未満のスケールとなっています。その範囲でも十分なデータと有効なアーキテクチャを用意すれば精度と汎用性を兼ね備えたポテンシャルを構築できるということを実験的に示したのがMLIPの成果のひとつかと思います。
このことは、計算コストの観点だけでなく、ある程度小規模なDFT計算で学習したMLIPを使って大規模シミュレーションを行うときの外挿性にも寄与しています。
古典ポテンシャルとグラフNNの対応関係については分子シミュレーション学会誌「アンサンブル」に寄稿した内容もあわせてご覧ください。 https://www.jstage.jst.go.jp/article/mssj/26/1/26_18/_article/-char/ja
局所近似が成立しない問題とその工夫
ここまで、MLIPがターゲットとしているポテンシャル関数というものが機械学習にとって特に都合の良い問題設定だったことを説明してきました。逆に言えば、この仮定から外れるケースではナイーブな局所近似が成立しづらいため、様々なアイデアが提案されてきています。
典型的には、MLIPの範囲を超えて電子状態に直接紐づく物性を予測したいといったケースがあります。たとえば、状態密度(DoS)やバンドギャップといった量はエネルギーとは異なり、非局所的な構造を強く反映しています。これらは機械学習の枠組みだけを見れば予測対象を変えただけではあるのですが、実際にはMLIPにおいて有効な近似は適用できないため、MLIPで有効なモデルが必ずしもそのまま適用できないという問題設定となっています。
MLIPにおいても、例えば電荷移動を伴う系では大域的な相互作用を含むため、局所性の近似が成立しづらくなります。より極端な例として電荷の総和がゼロでない系も考えられます。この場合も系全体での電荷割り当てという大域的な問題が発生します。(例えば、真空中にN個の分子が浮いているとしたとき、どの分子の電荷を変更すればいいのかについて解く必要があります)。
電荷移動を題材として、どういったモデルが現在考えられているのかを紹介しておきます。電荷分布それ自体に興味があるケースもありますが、ここではMLIPの文脈でとり上げます。古典ポテンシャルにおける標準的な解法は、QEq [1]のような緩和計算を導入して電荷移動を模式的に解く方法です。これを組み込んだMLIPとしては4G-HDNNP [2]が知られています。物理的解釈性に優れる一方で、ナイーブな実装では計算量は再びO(N^3)に上がってしまいます。そのため近年では、速度と精度のバランスをとる新たなアーキテクチャも提案されています [3, 4]。
別のアプローチとして、対象系を限定する方法もあります。たとえばOMol25のベースラインモデル [5]は孤立した分子系に対象を絞ることで、電荷割り当てを明示的に解かずに、全電荷がゼロでない推論を可能にしています。一方でこの方向性は、電荷やスピンの情報をモデルが直接的に扱える代わりに、局所性を仮定していたMLIPでは自然に確保できていた原子数方向の外挿性、示量性の問題が表に出てきます。
図1はOMol25データセットを学習したUMA (UMA-s-1p2 [6])を使い、NH4+分子の数を変化させたときの1分子あたりのエネルギーを示しています(紫色)。1分子のときの値をみると、電荷のないNH4とのエネルギー差は4.27 eVであり、これは実験値である4.698 eV [7]に近い値です。このため、UMAは単分子に対する電荷の影響を再現できると言えます。一方で、孤立したNH4+分子が複数ある場合は、本来は各分子がNH4+となることが自然であり、1分子あたりのエネルギーはNH4+と同等になることが期待されます。しかしながらUMAでは分子数に依存する結果となりました。これは、現在のUMAのスコープから外れて、電荷の扱いによって示量性が満たされなくなったことを示しています。
図ではまた、電荷のないNH4分子についても計算しています(緑色)。こちらは、NH4単分子ではdoubletラジカル(spin=2)とみなして計算し、複数分子の系では最大スピン状態(spin=N+1)に対応するようにしました。こちらも同様に分子あたりのエネルギーは一定になることが期待されますが、spinを明示的に設定するUMAでは分子数に対する依存性が出ています。
このように、モデルが取り扱うことのできる系の多様性と適用範囲の外挿性の間には繊細なトレードオフの関係があることがわかります。
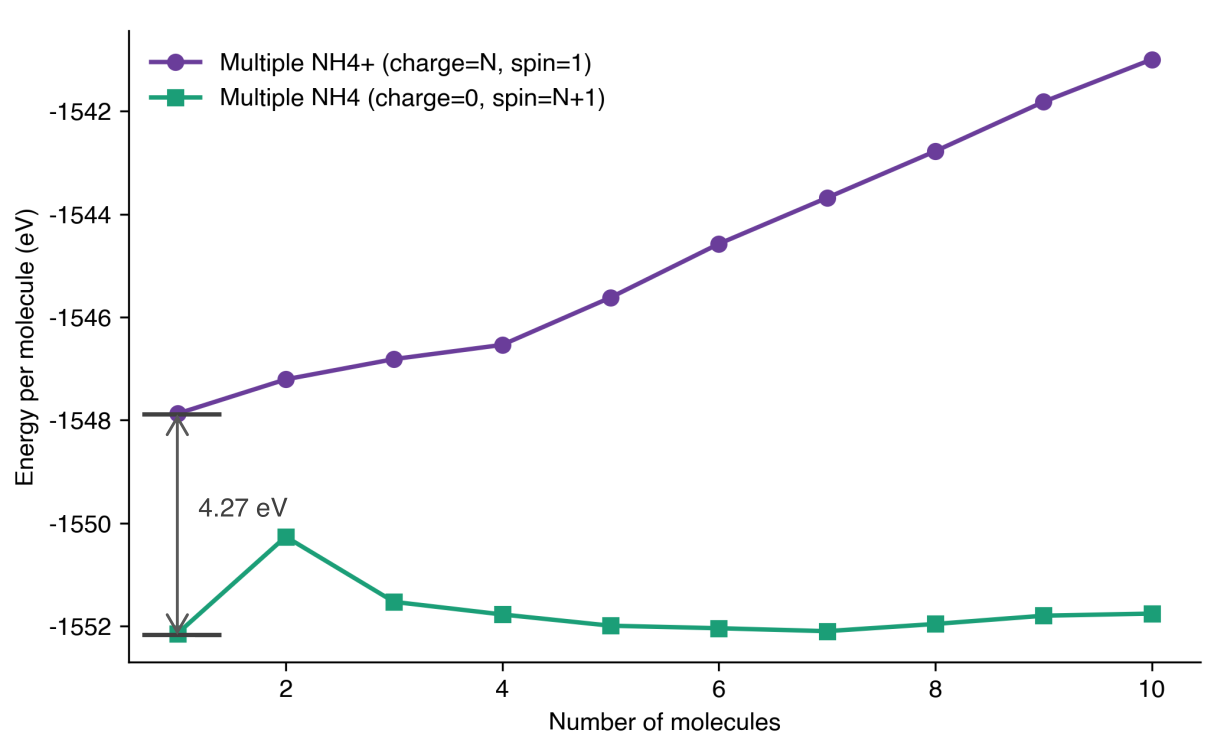
図1: UMA (UMA-s-1p2) を使ってNH4複数分子の系を推論したときの分子数と分子あたりのエネルギーの関係。Multiple NH4+の系列では、spinは1で固定して系全体の電荷を分子数Nと一致するように変化させて推論した。Multiple NH4の系列では、電荷は0としてspin=N+1とした。複数分子の配置は分子間の距離がそれぞれ10 nmとなるように直線状に配置した。構造最適化はASEライブラリのFIRE法を使い、力の収束条件を fmax=0.01 (eV/Å, 1 Å=0.1 nm) とした。
PFP開発においての私たちの今後の方針ですが、大きな方針としては任意の系にそのまま適用できる汎用性の追求を最重視しています。そのため、汎用性、原子数に対する精度、速度のスケーラビリティのいずれも妥協しないという観点でアーキテクチャの改良を進めていくことになります。方向性としては線形あるいはそれに準ずるスケールの計算量で大域的な相互作用を扱っていくことになると思います。
いずれにせよ、実用的な計算で性能が出ることがMLIPの価値となります。そのため、ベンチマークを起点とし、どの系でうまくいくのか、いかないのかを定量的に評価しながら技術を導入していくことになります。
私達Materialsチームでは現在、実用性を志向したPFPのベンチマークを拡充しています。
PFPを用いた格子熱伝導率計算
PFPを用いた表面エネルギーのベンチマーク
インターン募集の案内について
2026年もPFNでは材料開発にまつわる研究をテーマとしたインターンを募集しています。
https://www.preferred.jp/ja/careers/internship
私たちはこういった現行のMLIPでは取り扱いが困難と思われる問題設定に対しての先験的な研究を行っています。特にインターンではこうした新奇な研究テーマに対して積極的に取り組んでおり、いくつかのテーマは学会発表あるいはブログ記事等の形で公開しています。
PFN Materialsセミナーで、材料分野のPFN最新研究を紹介しました
スピン自由度を考慮したNeural Network Potentialの構築とcDFTを用いたデータセット作成
励起状態 Neural Network Potentialの開発
今回の記事はMLIPのアーキテクチャに特化した話題ですが、もちろんインターンではMLIPにとどまらず、様々な形での機械学習と材料科学の融合領域に関わるテーマを扱っています。以下は2025年に関連する発表です。他の記事はブログのタグから一覧を見ることができます。
拡散モデルを用いた結晶構造の対称性を考慮した条件付き生成
GNNに基づく凝集系における結合推定モデルの開発
自ら改善し進化する探索システム: 進化的探索エージェント
P-DRUM: Post-hoc Descriptor-based Residual Uncertainty Modeling for Machine Learning Potentials
基盤モデル開発というとどうしても大規模データと学習の「物量で殴る」イメージが先行しがちですが、今回紹介したようにMLIPの開発は工学的な課題の精緻化と実用面での価値の両睨みをしつつ物理から機械学習まで様々な知識を動員して開発していく営みでもあります。材料開発を通して社会に貢献できるよう、私たちはその境界を押し広げ続けていきます。
References
[1] A. K. Rappe and W. A. Goddard III, “Charge equilibration for molecular dynamics simulations”, J. Phys. Chem. 95, 3358-3363 (1991).
[2] Tsz Wai Ko, et al., “A fourth-generation high-dimensional neural network potential with accurate electrostatics including non-local charge transfer”, Nature Communications 12, 398 (2021).
[3] Ilyes Batatia, et al., “MACE-POLAR-1: A Polarisable Electrostatic Foundation Model for Molecular Chemistry”, https://arxiv.org/abs/2602.19411v1 (2026).
[4] Alessandro Caruso, et al., “Extending the range of graph neural networks with global encodings”, Nature Communications 17, 1855 (2026).
[5] Daniel S. Levine, et al., “The Open Molecules 2025 (OMol25) Dataset, Evaluations, and Models”, https://arxiv.org/abs/2505.08762 (2026).
[6] fairchem by the FAIR Chemistry team. https://github.com/facebookresearch/fairchem
[7] https://atct.anl.gov/Thermochemical%20Data/version%201.122p/species/?species_number=1051