Blog
PFNリサーチャーの石谷です。この投稿は2022年8月12日にJournal of Chemical Information and Modeling誌に出版された論文「Molecular Design Method Using a Reversible Tree Representation of Chemical Compounds and Deep Reinforcement Learning」についての解説記事です。論文はオープンアクセスで以下のURLから閲覧可能です。
https://pubs.acs.org/doi/10.1021/acs.jcim.2c00366
概要
より良い物性を持つ分子の自動設計は、in-silico創薬や材料設計の初期段階において重要なプロセスの一つです。本論文では、深層強化学習による新たな分子自動設計手法を提案しました。この手法では、新たに考案した分子の粗視化表現と深層強化学習を組み合わせることで、目的とする物性を最適化した分子を設計することを目指しました。この粗視化表現「Reversible Junction Tree (RJT)」は可逆的に元の分子に復元可能なため、強化学習による最適化をより効率的に行うことができると考えられます。実際、本手法「RJT-RL」を用いることで、単純なベンチマークタスクだけでなく、より実際の創薬応用に即したケースにおいても、効率的に所望の性質を持つ分子を自動設計できることがわかりました。
目次
背景
創薬や材料設計の初期ステップとして新規化合物の探索が重要ですが、この探索には実験の試行錯誤が必要なため費用と時間がかかるという点が問題になっています。一方近年、化合物の探索を計算機内で行うin silicoの創薬や材料探索が注目をあつめており、膨大な計算資源が利用できれば、実験だけでは困難であったより広い化学空間の探索が可能になると考えられています。しかしながら、化合物が持つ離散的な化学構造の空間の探索は一般的に困難であり、効率的な手法が求められています。そこで近年、ディープニューラルネットワーク(DNN)を用いた新たな探索手法、例えば変分オートエンコーダ(VAE)などの生成モデルを用いた手法が提案されてきました[1]。これらの手法は、DNNで近似された生成モデルによって化合物の離散空間と連続的な潜在空間との間のマッピングを学習させ、化合物の最適化をこの連続的な潜在空間で行えるようにすることで、この問題を回避しようとするものです。しかし、この方法にはいくつかの問題点がありました。例えば学習されたマッピングが実際効率的に最適化を行うのに適しているかどうか評価するための客観的な良い指標が存在しない点、生成モデルの学習過程が最適化ターゲットである分子のスコア関数に関する化合物の最適化過程から切り離されている点、などが挙げられます。
もう一つの例は、強化学習に基づく方法です。この方法では、分子設計をマルコフ決定過程と考え、エージェントが周囲の環境から提供される報酬に基づいて最適な方策を学習していきます。強化学習では、DNNモデルで近似された方策が最適化中に分子のスコア関数を学習するため、上記の潜在空間ベース法の問題を一部回避することができると考えられます。
これらのDNNを用いた手法は、入力に用いる分子の表現方法として、文字列表現(SMILESなど)を用いるものと、化学構造のグラフ表現を用いるものに大別できますが、いずれも問題点がありました。例えばSMILES表現を用いる方法は、生成されたSMILESに文法的に無効なものが含まれる可能性があり、これが最適化に影響を与えることが懸念されます。一方グラフ表現では、SMILES表現で起こる無効な分子の問題は部分的に回避できますが、正しい原子価を満たすかどうかや、芳香環がヒュッケル則を満たすかどうか、さらにはdrug-likeな化合物を効率的に学習できるかなどの問題点が依然存在していました。
このような問題を解決する手法として、化学構造のグラフの木分解を利用した新しい粗視化分子表現法(Junction Tree; JT)が提案されました[2]。このJT表現では、分子は環や結合に対応するノードを持つ木(閉路を持たないグラフ)として表現され、常に有効な分子を表現できます。しかし、JT表現は補助的な情報なしに元の分子に逆変換することができないため、実装が複雑になり計算コストがかかるという問題が残っていました。
そこで本論文では、まず、JT表現を拡張することで、補助的な情報なしに元の分子に逆変換できる可逆JT (RJT)表現を提案しました。そしてこのRJT表現を用いた深層強化学習ベースの分子生成・最適化フレームワークを提案しました。
方法
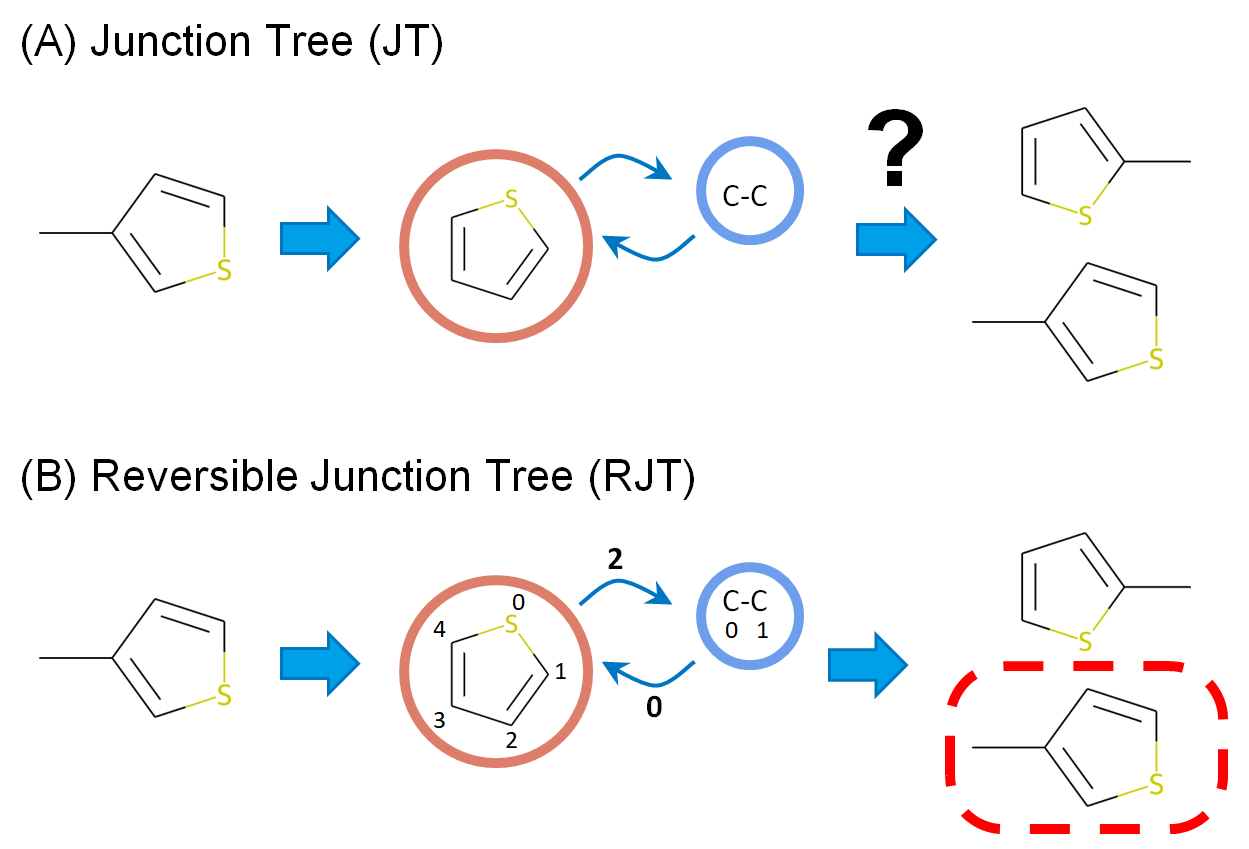
図1: RJTの概念図
まず、本方法のベースとなるJT表現について、詳しく見ていきたいと思います。JT表現の問題は、上述のように、不可逆であり元の分子に戻せないという点でした。例えば、元の構造が環構造を含む場合、環は1つのノードとして表現され、それに結合した置換基は、そのノードに結合したノード(あるいはサブツリー)として表せます(図1A)。一方、これを元の化合物に戻そうとすると、環のどの位置に置換基が結合していたか情報が失われており、もとに戻すことができません。
そこで、本方法では、環のどの原子上に結合していたかの情報(サイト情報)をJT表現のツリー構造のエッジに保持させるようにしました(図1B)。JT表現から元分子に戻す際には、このサイト情報を参考に、環とボンド(あるいは環と環)を結合させていけば、復元することが可能です。以降、この拡張したJT表現を、RJT表現と呼ぶことにします。
このRJT表現を深層学習で使用可能にするためには、RJTを固定長の隠れベクトルにエンコードする必要があります。JT表現の場合[2]では、同様のエンコードに、木構造に適用可能なように拡張されたGRU (TreeGRU)が用いられていましたが、ここではさらに、このTreeGRUにノードだけでなくエッジの特徴量も埋め込むことができるようにする拡張を行いました。このRJT用のDNNをRJTNNと呼ぶことにします。
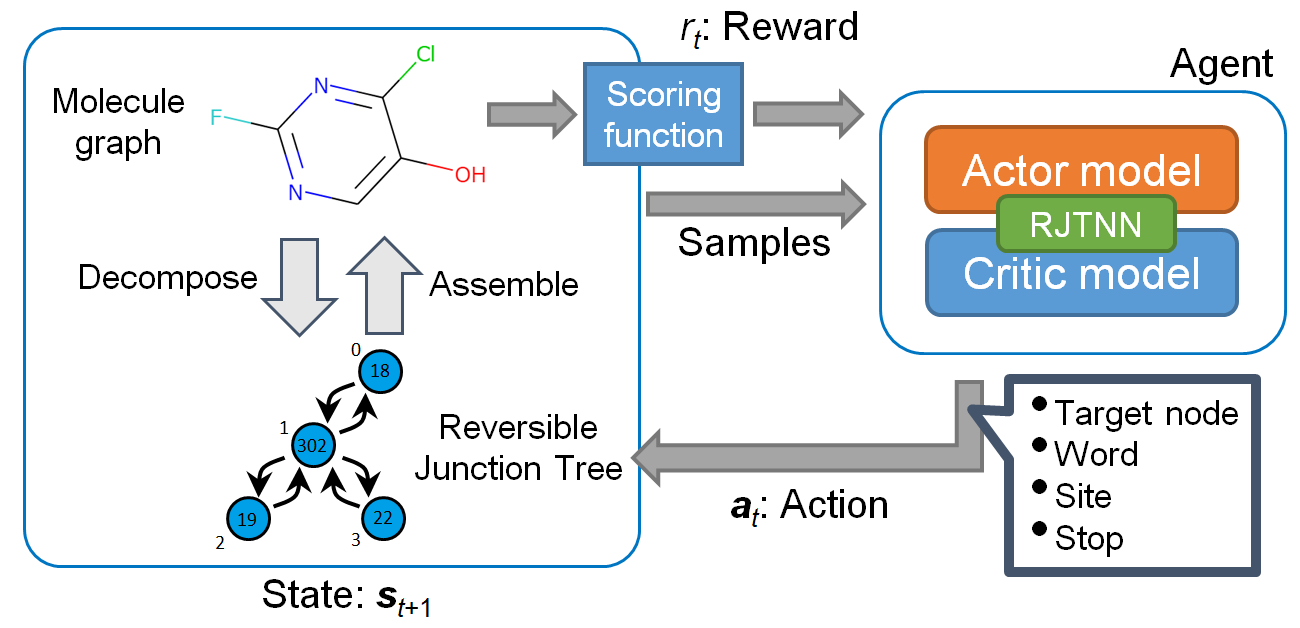
図2: RJT-RLの概念図
次に、分子の生成をRJT表現の木構造を生成するタスクと考え、このRJTを強化学習で生成することを考えました(図2)。RJT表現は一対一で分子構造に対応するため、強化学習の報酬としてこの分子から計算できるスコア関数を使用すれば、目的の物性が高いと予測される分子を生成できると期待されます。強化学習のアルゴリズムとしてproximal policy optimization (PPO)を使用しました[3]。このRJTとPPOを使用した分子生成法「RJT-RL」では、エピソードが完了後に出力されたRJTのみならず、強化学習の各ステップで生成された中間状態に対応するRJTも基本的に分子にデコード可能なため、全て報酬が計算可能です。このように途中状態に対しても報酬を与えることで、より効率的に最終的なスコアが高くなるような方策を学習できるようになると期待されます。これは、本方法がREINVENT[4]など従来のSMILESベースの強化学習手法と異なる点の一つです。
また、強化学習では、探索と利用のバランスが重要であり、利用に行動が偏ると局所的な最適状態に陥って最適化が進まなくなる問題が起こります。オリジナルのPPOの実装にも、探索を促進するためエントロピー項が導入されていますが[3]、RJT-RLではさらに探索を促進するため、duplication penaltyを導入しました。これは、同じ分子を指定回数以上生成した場合に、報酬をゼロにするようなペナルティを与えるものです。
本手法の有効性を検討するために、いくつかのテストケースで既存手法を含め性能を比較しました。特に、強化学習の中間状態に対しても分子のスコア関数を評価するケース(step reward)と強化学習エピソードの終了時のみに評価するケース(final reward)、さらには、上述のduplication penaltyを用いるケースと用いないケースなど、いくつかの組み合わせを用いて評価を行いました。
さらに、本手法と既存手法を比較するため、SMILESベースの深層強化学習を用いた手法であるREINVENT[4]と、フラグメントベースかつルールベースの手法であるCReM[5]を取り上げました。REINVENTは事前学習が、CReMはフラグメントライブラリが必要となりますが、条件をそろえるため、いずれもzinc250k[6]をデータセットとして用いました。
詳しい方法とアルゴリズムについては、本論文を参照してください。
結果
Penalized LogPをターゲットとした最適化
まず、化学構造のみから容易に計算できるスコア関数を用いて、本手法の有効性を検証しました。最初の実験では、水-オクタノール分配係数(LogP)に環サイズや合成可能性(SA)も考慮したPenalized LogPスコア[7]をターゲットとした最適化が行えるかどうかを評価しました。実験条件としては、step rewardでduplication penalty無し(P1)、final rewardでduplication penalty有り(P2)、step rewardでduplication penalty有り(P3)、の 3 つのケースを比較しました。すべてのケース(P1, P2, P3)において、duplication penaltyのある場合とない場合どちらでも、高い報酬を得られる分子が連続的に生成されていることが分かります(図3A-C)。Final reward (P2)とstep reward (P1, P3)を比較すると、P1とP3ではP2よりも報酬関数が早く上昇する傾向が見られました(図3A-C)。この結果は、中間状態の報酬が最適化プロセスを促進することを示唆しています。P4のREINVENTにおいても、高スコアの分子を連続的に生成できていますが、そのスコアはRJT-RLで生成したものを上回りませんでした(図3D)。
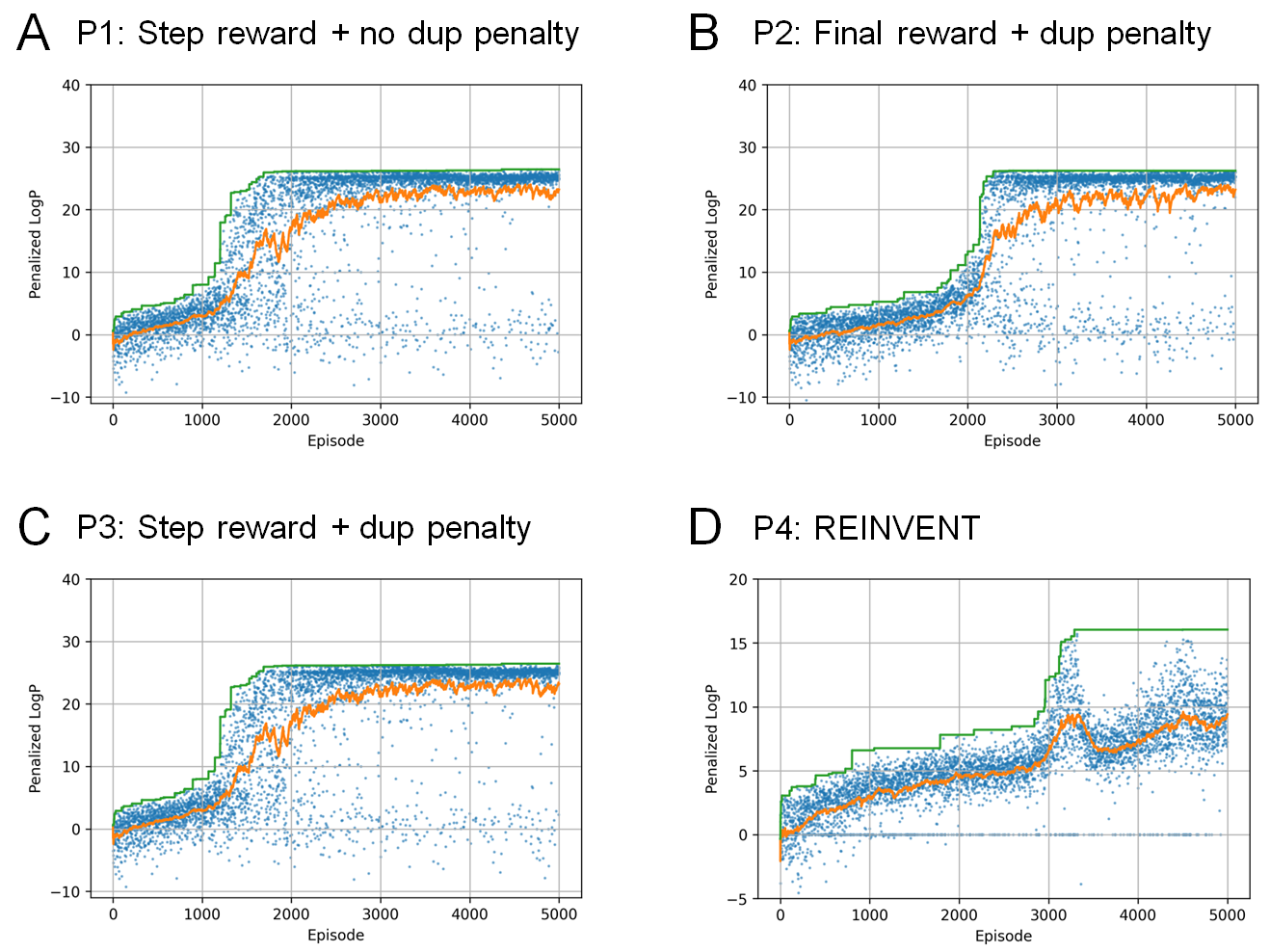
図3: Penalized LogP実験の結果
実験で得られた上位3つのペナルティ付きLogPスコアを、先行研究の結果とともに表にまとめました。Penalized LogPが高い物質をデザインすること自体に、実用上の意味はありませんが、潜在空間ベースの最適化法では、学習データにないような高いLogPを持つ物質を生成できません。一方で、最適化過程で生成モデルを目標関数に従って更新するweighted retraining[8]では高いLogPを持つ分子を生成できることが分かっています(参考)。この比較から、RJT-RLは、weighted retrainingと比較しても遜色ない性能を発揮することが分かりました。
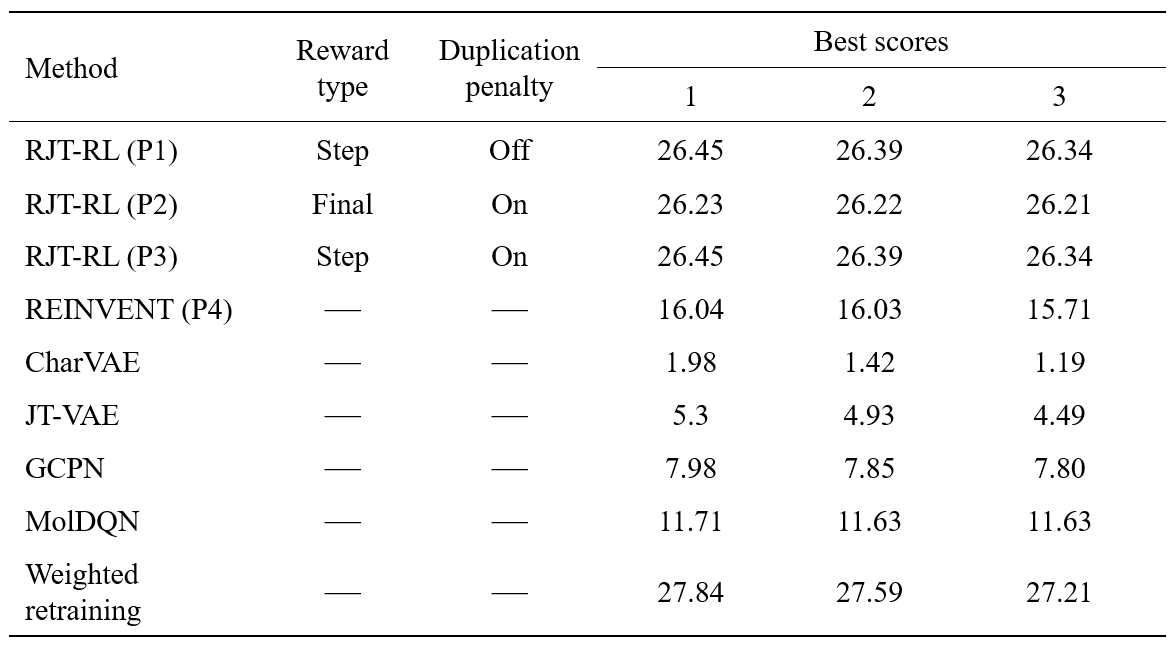
表: RJT-RLと他手法の比較
Fingerprint類似性をターゲットとした最適化
次に、指定したターゲット化合物に類似した分子を生成するという問題設定で、本手法の有効性を検証しました。ここでも、報酬設定の違いによる効果を評価するため、Penalized LogPのケース同様に、RJT-RLの3つのケースとREINVENTを比較しました。さらに、ルールベースの手法としてCReMを比較しました。ターゲット化合物としては、Vortioxetine (図4A; 結果はS1-S5)とCelecoxib (図5A; 結果はC1-C5)を用いました。
実験の結果、RJT-RLにおいては、duplication penalty + step reward (S3, C3)のケースでほぼ完全一致(similarity=1.0)という高い類似性の分子が得られることがわかりました(図4D、図5D)。特に、duplication penaltyなしの場合はある程度最適化が進んだところで局所最適解から抜け出せなくなり、同じ化合物が生成され続けほとんど探索が進まないのに対し(図4B、図5B)、duplication penaltyありの場合では探索が進むことから(図4C, D、図5C, D)、効率的な探索にduplication penaltyが重要であることが分かりました。一方でREINVENT (S4, C4)では、類似性がやや改善されるものの、大きく改善することはできない結果となりました(図4E, 図5E)。特にcelecoxibについては、REINVENTの原著論文[4]では完全一致の化合物に到達できていたため、この違いは事前学習に用いたデータセットの違いに起因するものと考えられます。つぎにCReMでは、celecoxibについては完全一致(similarity=1.0)の化合物に到達できることがわかりました(図4F, 図5F)。また、vortioxetineでは、完全一致ではないが、類似性の高い化合物に到達できています。CReMは分子生成が高速であるため、評価関数が単純な今回のケースでは、RJT-RLよりも高速に最適解に到達できました。ただ、スコアの評価回数(図4, 5の横軸)で比較すると、CReMのほうがRJT-RLに比べて多数の評価が必要となることがわかります。
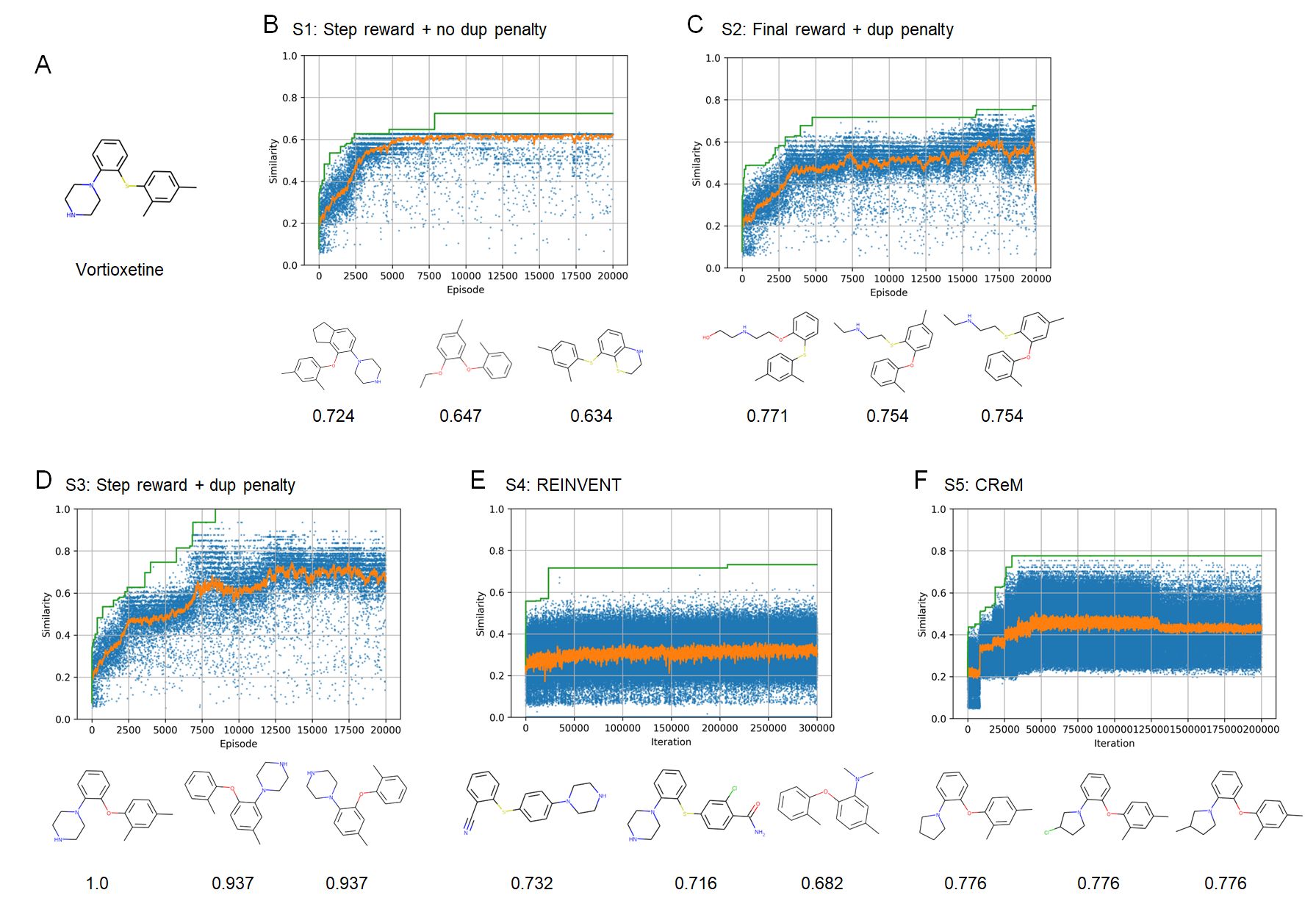
図4: Vortioxetineをターゲット分子とした最適化実験の結果
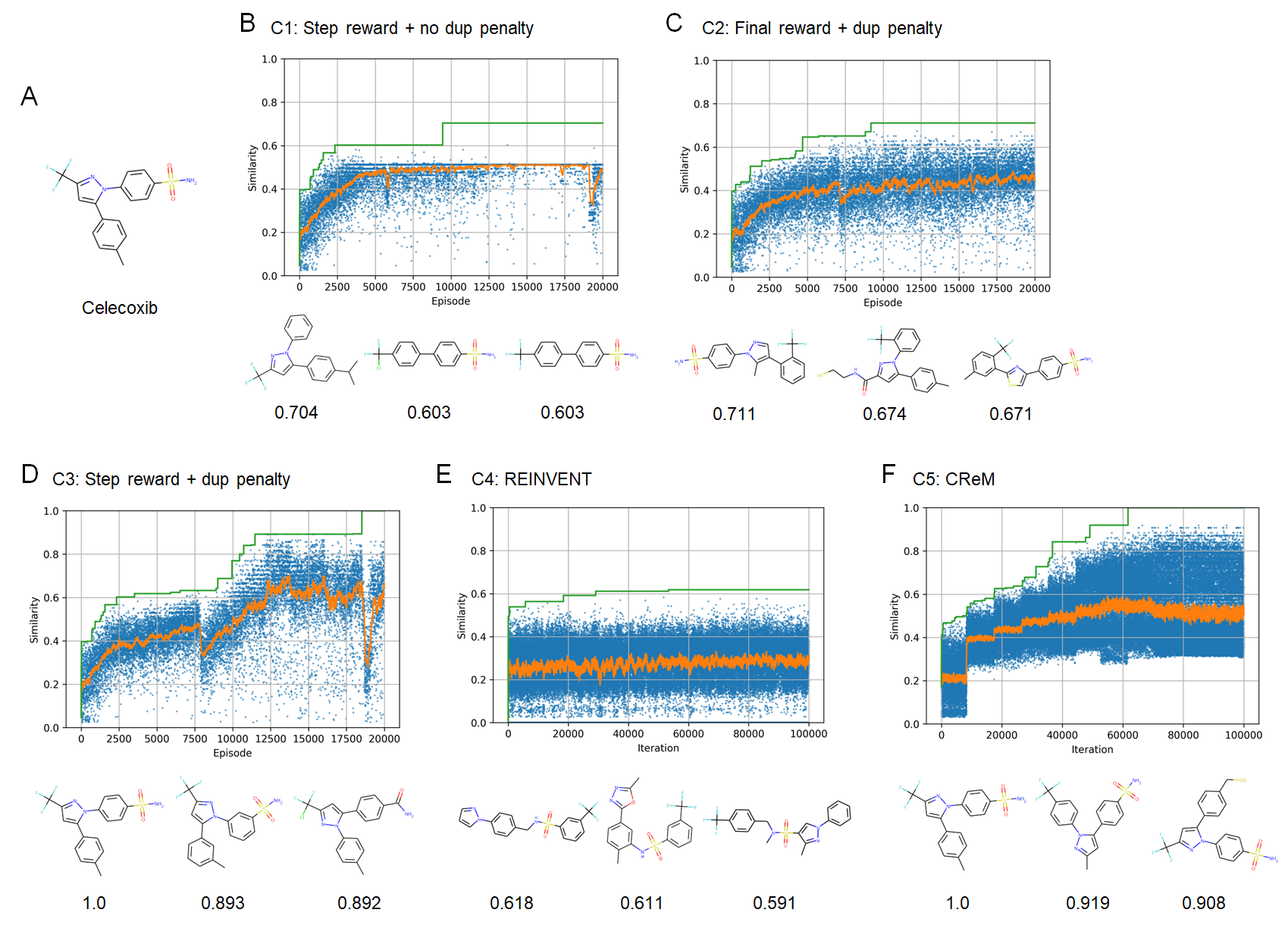
図5: Celecoxibをターゲット分子とした最適化実験の結果
構造ベースのスキャフォールド・ホッピング
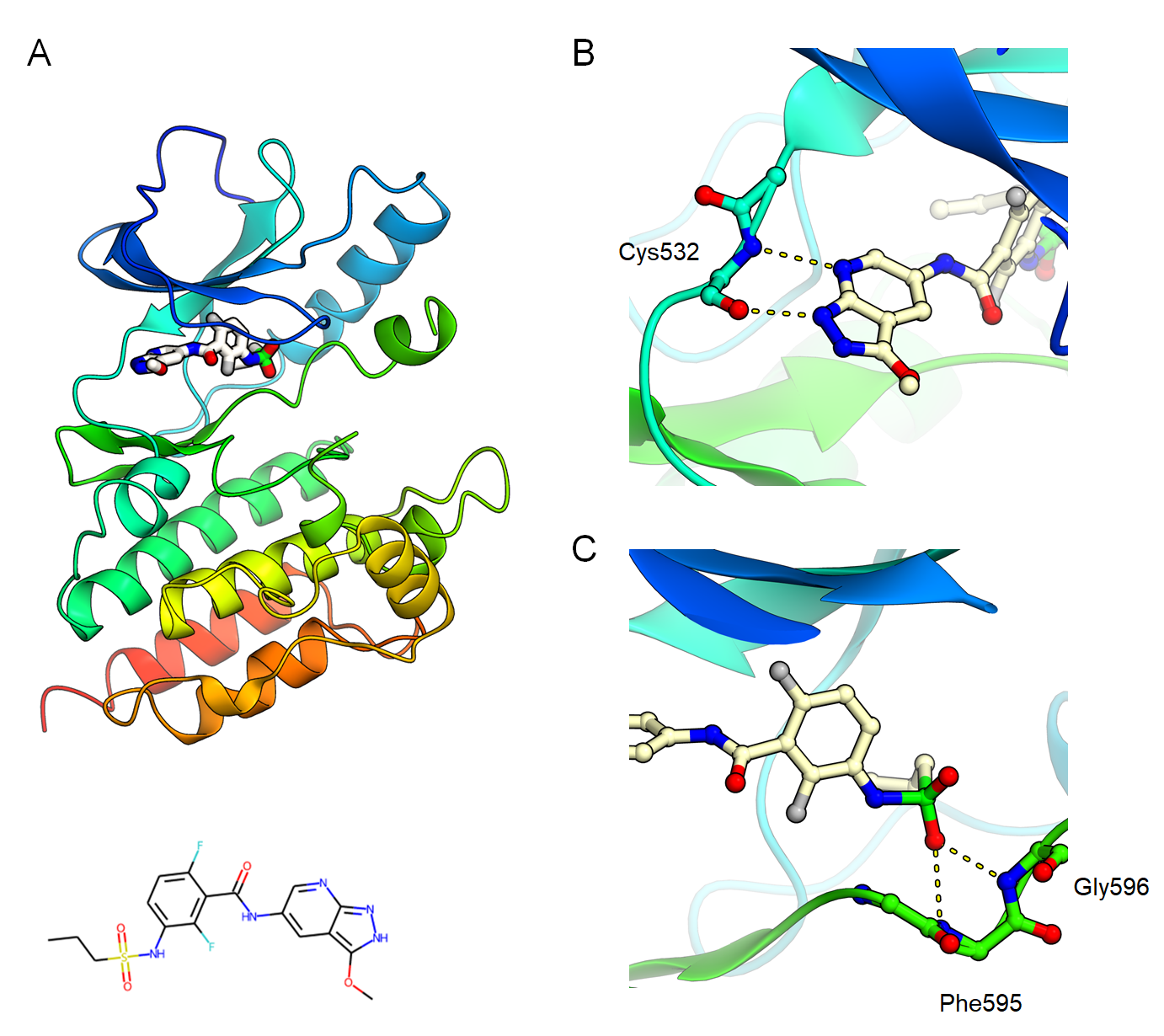
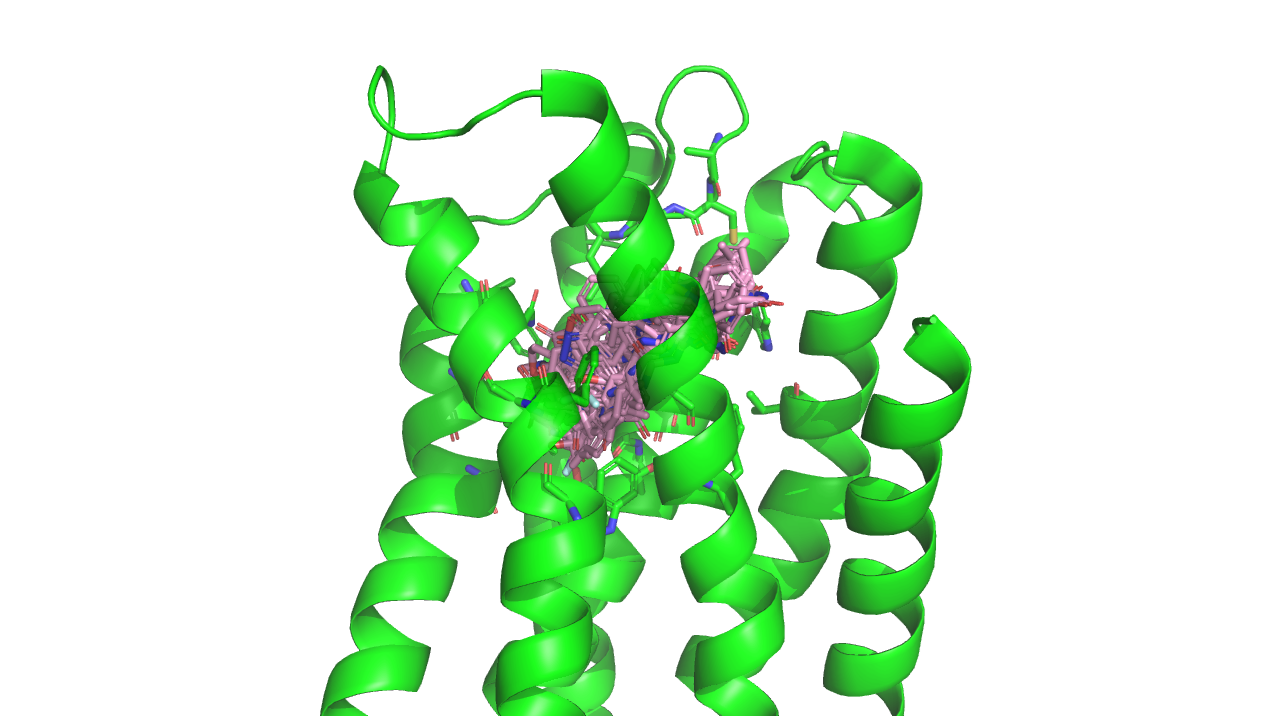
図6: B-Rafと阻害剤複合体の立体構造
次に、より実応用に近い問題設定として、構造ベースのスキャフォールド・ホッピングを取り上げました。実際の構造ベース創薬応用では、標的タンパク質との既知の重要な相互作用を保持する異なるスキャフォールドを持つ分子を探索することが重要です。ここでは、
B-Rafキナーゼとその阻害剤の複合体構造[9](図6A) を例として取り上げました。この構造では、阻害剤のピリミジン窒素がキナーゼのヒンジ領域のGly 596の主鎖窒素原子と水素結合し(図6B)、さらに、スルホンアミド基を介してCys 532の主鎖窒素原子と相互作用しています(図6C)。そこで、ヒンジ領域とピリミジン基との相互作用はそのままに、Cys 532との相互作用点を変化させた新規化合物の設計を試みました。
強化学習の報酬として用いるスコア関数は以下のように定義しました。
\[
R_{\mathrm{fin}}=\mathrm{Intr}\cdot c_{\mathrm{intr}}-\Delta G \\
\mathrm{Intr} = \mathrm{Intr}\left(\mathrm{G596}-\mathrm{N}, \mathrm{HBA}\right) + \mathrm{Intr}\left(\mathrm{C532}-\mathrm{N}, \mathrm{HBA}\right) \\
\]
ここで、\(\mathrm{Intr}(\cdot, \cdot)\)は相互作用を評価する関数、HBA は化合物の水素結合受容体原子、\(\Delta G\)は化合物のドッキングポーズの AutoDock Vina スコア[10]、\(c_{\mathrm{intr}}\) はハイパーパラメータです。
相互作用関数は以下のように定義しました*1。
\[
\mathrm{Intr}\left(p, c\right) = \left\{
\begin{array}{ll}
1-\frac{d(p, c)-d_{\mathrm{min}}}{d_{\mathrm{max}} – d_{\mathrm{min}} } & (d_{\mathrm{min}} \lt d(p,c) \leq d_{\mathrm{max}})\\
0 & (d_{\mathrm{max}} \lt d(p,c))\\
1 & (d(p,c) \leq d_{\mathrm{min}})
\end{array}
\right.
\]
ここで、\(d(p,c)\)はタンパク質と化合物の指定された原子間の距離とします。本実験では、\(d_{max}=6.0\)Å、\(d_{min} = 3.0\)Åを使用しました。
ドッキングスコアの計算に当たっては、生成された化合物の構造式をもとにRDKitで3次元配座を生成したのちドッキングシミュレーションを行って、スコア計算を行いました。RJT-RLで生成される化合物は、立体異性を考慮していないため、RDKitで可能な立体異性を全列挙するケースと、ランダムで選択するケースで比較実験をしました。すなわち、RJT-RLについては、final rewardを用いるケース (D1)、step rewardを用いるケース (D2)、final rewardを用いかつ立体異性を列挙するケース (D3) を比較しました。さらに、他手法として、類似性スコアで性能を発揮したCReM (D4)を比較しました。
実験の結果、いずれのケースも、相互作用スコアと、ドッキングスコア両方を改善するような化合物が生成できることがわかりました(図7)。D1 (final reward)に比べD2 (step reward)の方が報酬の上昇が早いため、より効率的に最適化が行われていると考えられますが(図7A, B)、実際には1分子生成するために多数のドッキングシミュレーションが実行されるため、時間でみるとD1 (final reward)の方が高速に最適化できていることになります。また、D3 (立体異性を列挙するケース)も、より報酬の上昇が早いですが(図7A, C)、こちらも1分子生成するために多数のドッキングシミュレーションが実行されるため、時間でみるとD1 (final reward)の方が効率が良くなります。ただ、このD3のケースでは多数のdocking計算は全て並列実行可能なため、各立体異性体に対する計算を並列化することで高速化が可能と考えられます。
一方、CReM (D4)では、報酬全体でみると改善しているものの、スコアごとに見ると、ドッキングスコアはRJT-RLよりも大きく改善している一方で、相互作用スコアがあまり改善出来ていないことが分かります(図7D)。CReMでは、このように2つの項が不均衡に最適化されてしまう問題があることが分かりました。
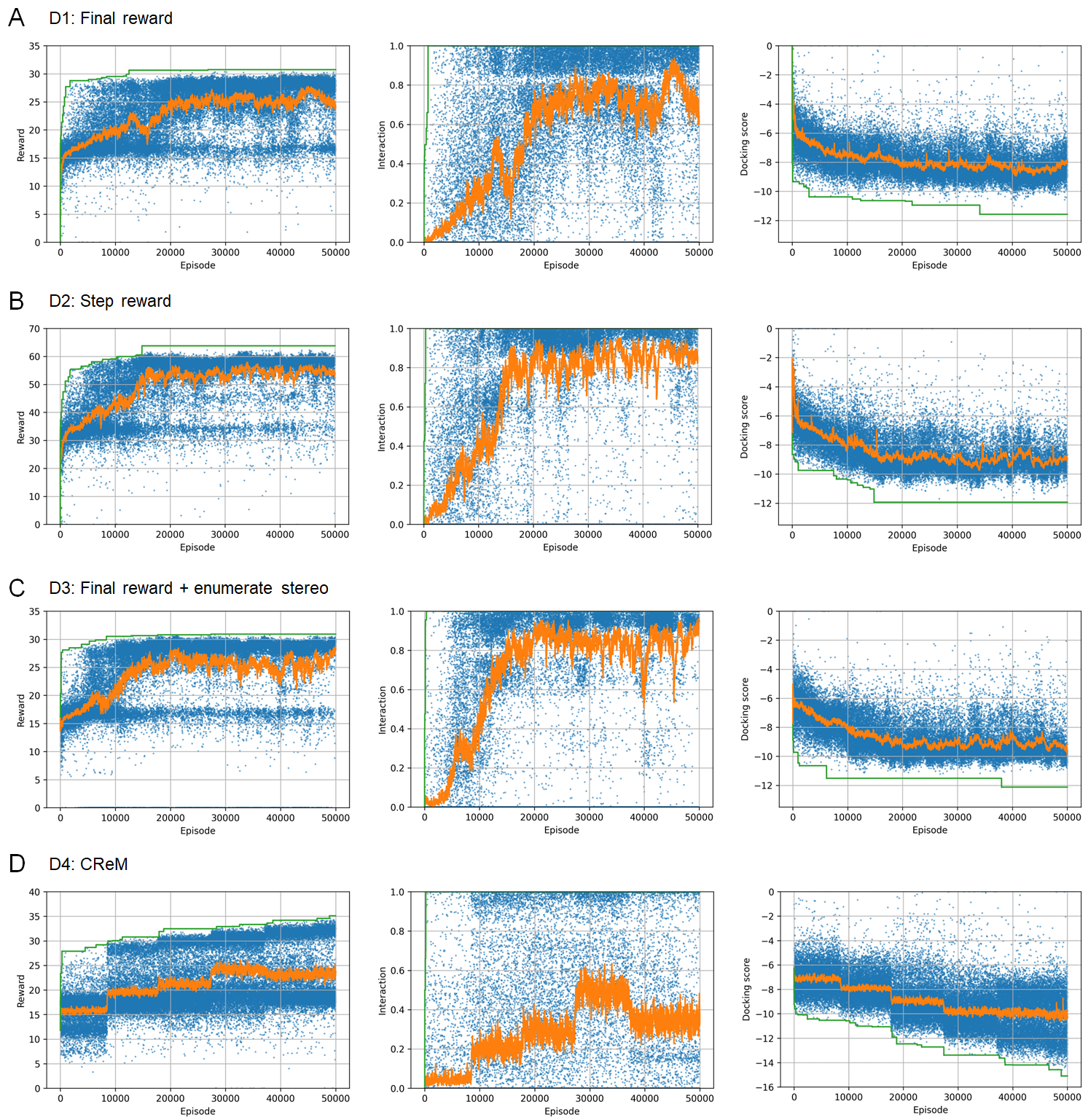
図7: 構造ベースのスキャフォールド・ホッピングの結果
多目的最適化
さらに複雑なケースの例として、多目的最適を取り上げました。実際の創薬シード探索においては、化合物のターゲットに対する相互作用だけでなく、化合物自体の物性も重要となってきます。上記のスキャフォールド・ホッピングの例に加え、さらに脂溶性 (LogP)と合成容易性(SA)スコアの範囲を指定範囲(\(\mathrm{SA}<4, 0<\mathrm{Log}P<5\))に制限する項を追加し、このような多目的のターゲットを最適化可能かどうか実験を行いました。強化学習の報酬として用いるスコア関数は以下のように定義しました。
\[
\begin{split}
R_{\mathrm{fin}} &= \mathrm{Intr}\cdot c_{\mathrm{intr}} – \Delta G – p_{\mathrm{SA}}\cdot c_{\mathrm{SA}} – p_{\mathrm{Log}P}\cdot c_{\mathrm{Log}P} \\
R_{\mathrm{step}} &= (7-\mathrm{SA})\cdot c_{\mathrm{SA}} – p_{\mathrm{Log}P}\cdot c_{\mathrm{Log}P}
\end{split}
\]
ここで、\(p_{\mathrm{SA}}\)と\(p_{\mathrm{Log}P}\)は以下のように定義されます。
\[
\begin{split}
p_{\mathrm{SA}} &= \mathrm{max}(\mathrm{SA}-4, 0) \\
p_{\mathrm{Log}P} &= \mathrm{max}(\left|\mathrm{Log}P-2.5\right|-2.5, 0)^2
\end{split}
\]
D1-D3実験の結果を踏まえ、ドッキングスコアの計算には時間がかかるため、final rewardでのみdockingを考慮することとしました。そしてstep rewardでは容易に計算できるスコア(LogPとSAスコア)のみを計算するという構成になっています。
実験の結果を図8に示します。RJT-RL (M1)では、ドッキングスコア、相互作用スコアだけでなく、LogPとSAスコアの項も最適化され、指定された特性を持つ化合物が生成できていることが分かります(図8A)。CReMでも、同様のスコア関数を用いて最適化実験を行いました (M2)。その結果、D4 (LogPやSAスコアによるペナルティーを含まないケース)と同様、全体としてスコアは上昇するものの、dockingスコアの項のみ最適化され、所望の物性を持つ化合物が得られにくい、ということが分かりました(図8B)。
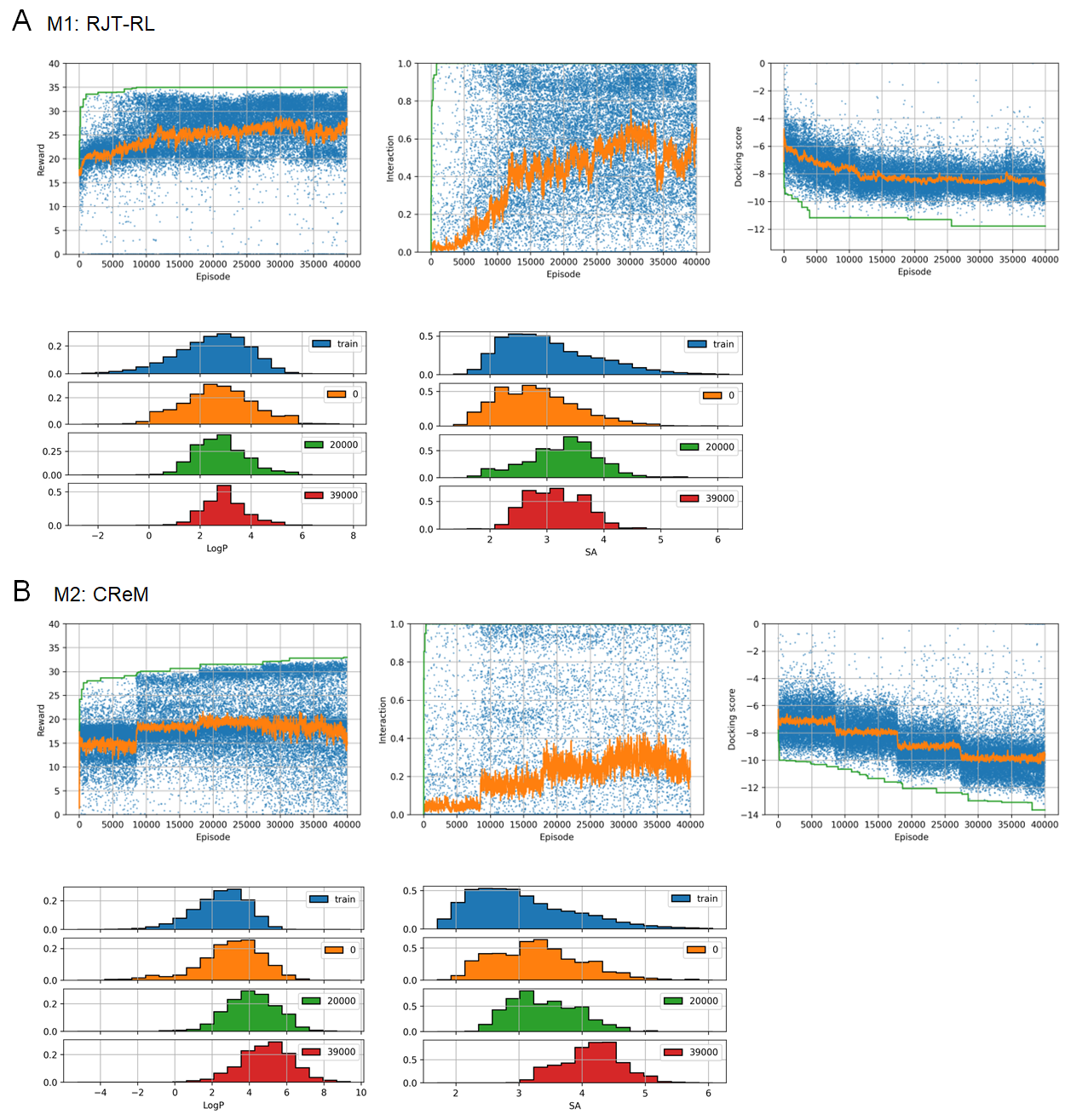
図8: 多目的最適化の結果
方策と価値関数のファインチューニング
最後に、DNNなど学習ベースの手法の利点の1つとして、あるタスクで学習したモデルを、他のタスクで転移学習やファインチューニングすることが可能である、という点が挙げられます。そこで,D1で学習したRJT-RLのモデルを、M1で定義された多目的報酬関数に対してファインチューニングを行う実験を行いました。
実験の結果を図9に示します。強化学習の初期のエピソードから、エージェントは高いドッキングスコアと相互作用スコアを持つ分子を生成できている事が分かります。これらのスコアはRL training中も高い状態で維持されています(図9A-C)。一方、SAスコアとLogPスコアは、学習の初期は指定範囲からはずれたものが生成されていますが、学習が進むにつれて徐々に向上し、指定範囲(\(\mathrm{SA}<4, 0<\mathrm{Log}P<5\))の化合物が生成されるようになってきています(図9D,E)。モデルのファインチューニングは、ターゲットタンパク質に対する相互作用など学習に時間がかかる報酬に対して学習した後に、様々な分子自体の物性の最適化を組み合わせて試す場合に、より短期間で評価ができるメリットがあると考えられます。
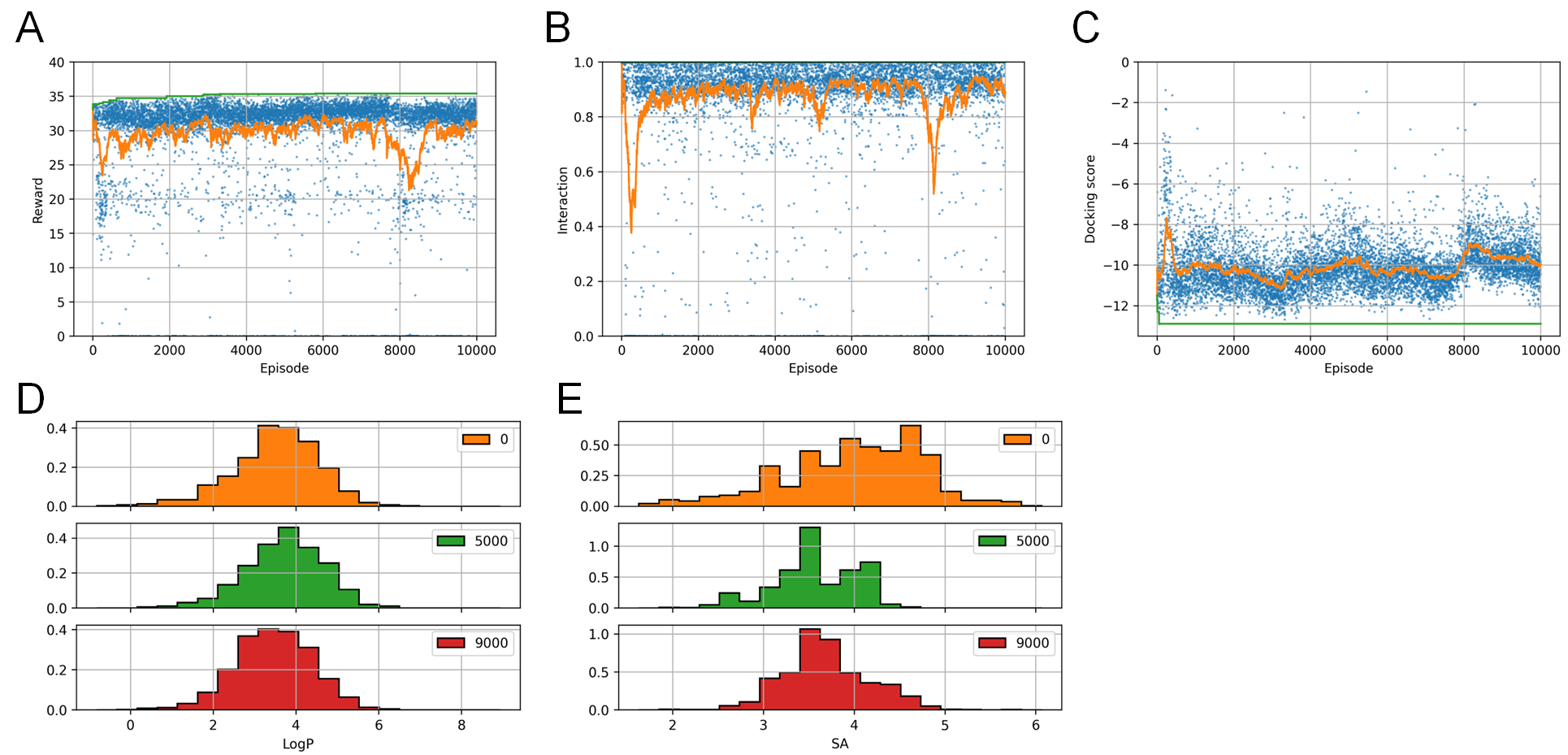
図9: ファインチューニングの結果
まとめ
本論文では、可逆的な分子の粗視化表現であるRJTを導入し、このRJTと強化学習を用いた新規の分子デザイン手法RJT-RLを提案しました。RJT-RLは、単純なベンチマークタスクにおいて、他の手法と同等あるいはそれ以上の性能を示すことがわかりました。さらに、多目的報酬関数を用いた構造ベースのスキャフォールド・ホッピングなどの、実応用に即したケースでも性能を発揮できることがわかりました。一方、本論文では、比較的小さなサイズのフラグメントから出発したde novoの化合物設計のみを考慮しましたが、ある程度性質が既知のリード化合物の最適化も化合物探索における重要なタスクの一つです。RJT-RLは現在RJTに対するノードの追加アクションにしか対応していませんが、リード化合物の最適化に適用するためには、さらにノードの削除や変更を含むように拡張していく必要があると考えられます。
*1: 論文中の報酬の定義式が誤っていましたので、訂正します。
参考文献
[1] Bilodeau C, et al. (2022) Generative Models for Molecular Discovery: Recent Advances and Challenges. WIREs Comput Mol Sci https://doi.org/10.1002/wcms.1608
[2] Jin W, et al. (2018) Junction Tree Variational Autoencoder for Molecular Graph Generation. arXiv 1802.04364 https://doi.org/10.48550/arXiv.1802.04364
[3] Schulman J, et al. (2017) Proximal Policy Optimization Algorithms. arXiv 1707.06347 https://doi.org/10.48550/arXiv.1707.06347
[4] Olivecrona M, et al. (2017) Molecular De-Novo Design through Deep Reinforcement Learning. J Cheminform 9:48. https://doi.org/10.1186/s13321-017-0235-x
[5] Polishchuk P (2020) CReM: Chemically Reasonable Mutations Framework for Structure Generation. J Cheminform 12:28. https://doi.org/10.1186/s13321-020-00431-w
[6] Gómez-Bombarelli R, et al. (2018) Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Central Science 4:268–276. https://doi.org/10.1021/acscentsci.7b00572
[7] Kusner M J, et al. (2017) Grammar Variational Autoencoder arXiv 1703.01925 https://doi.org/10.48550/arXiv.1703.01925
[8] Tripp A, et al. (2020) Sample-Efficient Optimization in the Latent Space of Deep Generative Models via Weighted Retraining. arXiv, 2006.09191. https://doi.org/10.48550/arXiv.2006.09191
[9] Wenglowsky S, et al. (2011) Pyrazolopyridine Inhibitors of B-Raf(V600E) Part 1: The Development of Selective Orally Bioavailable and Efficacious Inhibitors. ACS Med Chem Lett 2:342–347 https://doi.org/10.1021/ml200025q
[10] Trott O, Olson A (2009) AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function Efficient Optimization and Multithreading. J Comp Chem 30:2562–2574 https://doi.org/10.1002/jcc.21334