Blog
2021.09.13
Discovery of SARS-CoV-2 protease inhibitors using AI drug discovery technology
Tag
Kentaro Rikimaru
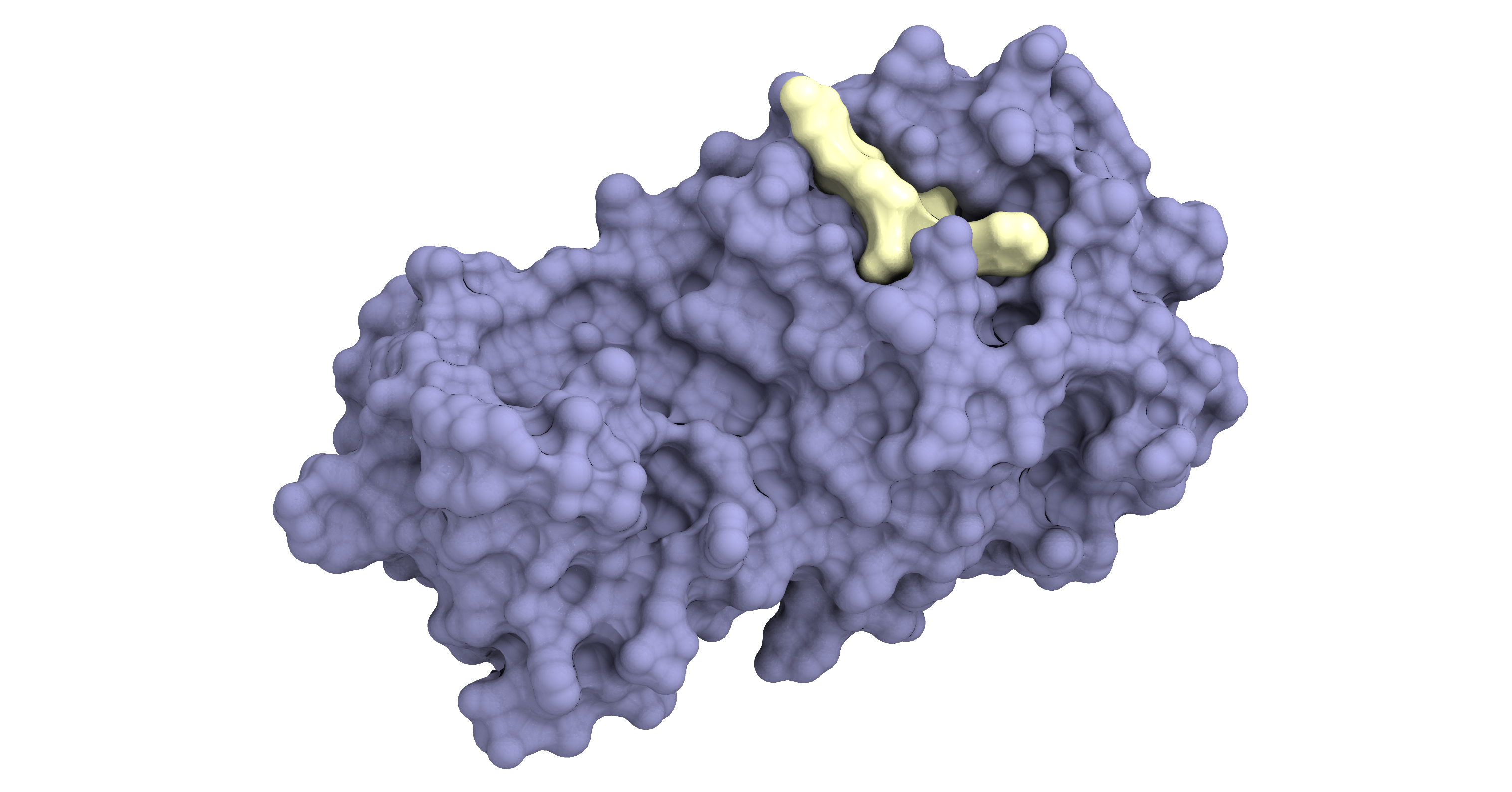
A pharmaceutical agent (in yellow) binding to the main protease of SARS-CoV-2 (in purple) to inhibit viral replication.
Introduction
The novel coronavirus disease (COVID-19) is currently expected to be controlled by vaccination, but the development of therapeutic agents targeting SARS-CoV-2 is highly desired to prevent pandemic recurrence due to further possible mutations in the virus.
Some therapeutic agents have been reported and approved for some clinical effects, but only for limited use: examples include direct injection of compounds targeting nucleic acid synthesis systems such as remdesivir, and antibodies to viruses such as REGN-COV2. Besides, many existing drugs have been studied for drug repositioning, but have not been approved yet. In addition, it is also desired to cope with the threat to mutant viruses by developing various therapeutic agents for COVID-19.
3CL protease (main protease, Mpro) is a protein essential for viral replication, and no highly homologous enzyme has been found in humans, making it a promising target for therapeutics in viral diseases. For example, rupintrivir is known as an Mpro inhibitor of rhinovirus, but it has not been applied to a therapeutic agent for COVID-19. Furthermore, most of the previous studies for Mpro inhibitors are focused on peptide-like compounds with leucine or similar structures, but the discovery of lead compounds with different structures is desirable to prepare for future viral mutations.
PFN has found Mpro inhibitors as candidates for the treatment of COVID-19 with a unique structure through a joint research with Professor Kenichi Akaji and Associate Professor Kazuya Kobayashi of Kyoto Pharmaceutical University, who have been conducting research on Mpro inhibitors of coronaviruses such as SARS virus and MERS virus for many years.
PFN’s AI drug discovery platform
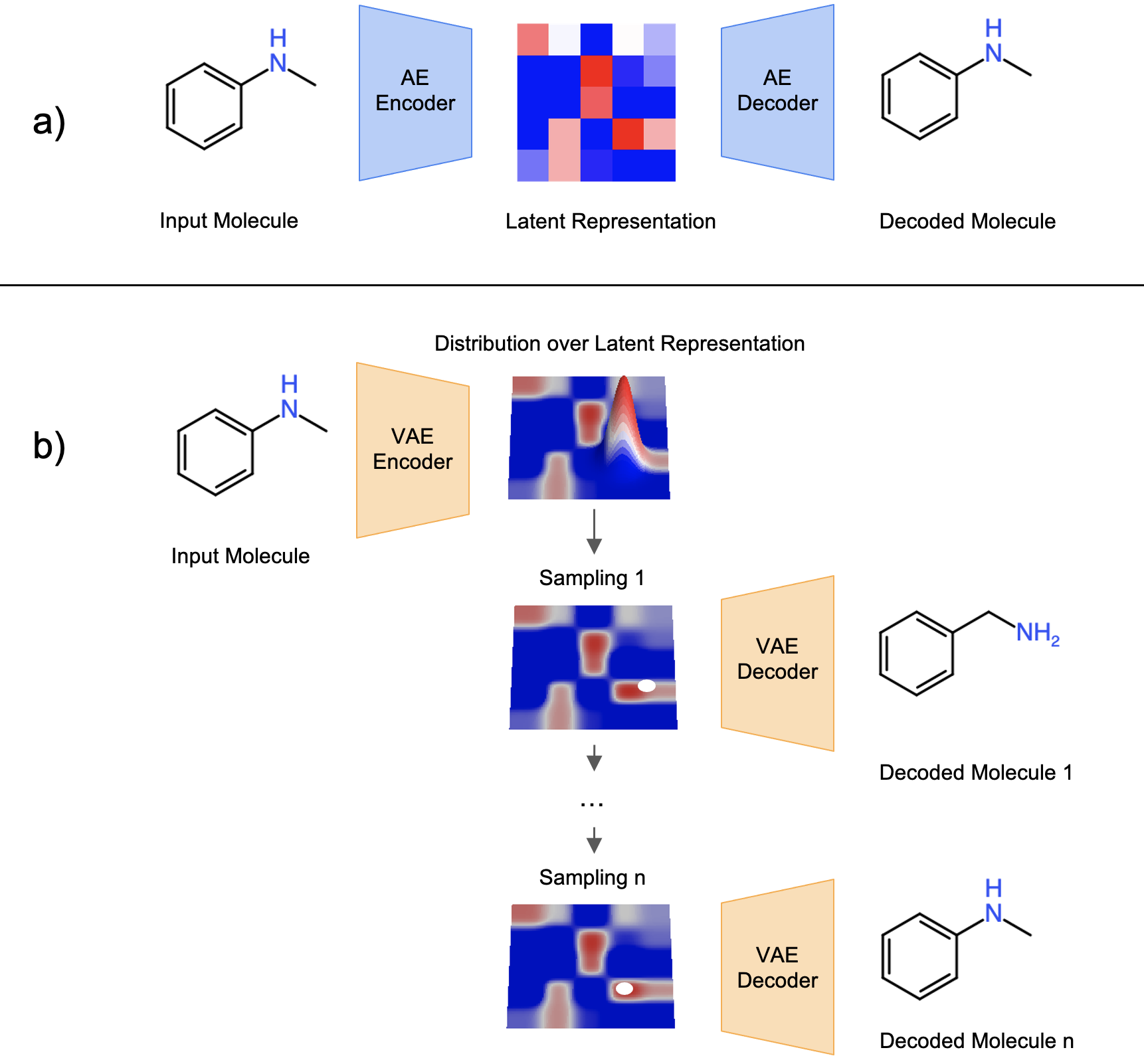
In recent years, image recognition and data generation using machine learning has made remarkable progress. Research that applies this method to pharmaceutical compound design (so-called AI-driven drug discovery) is getting a lot of attention. PFN is leveraging its deep learning expertise and abundant computing power to build its own AI drug discovery platform. This platform contains not only virtual screening using compound libraries designed by manual or rule-based structural modification, but also various compound generation models with optimization algorithms such as those implemented in Optuna to design and optimize compounds. It is possible to propose structures that are not included in the compound library or structures that are difficult to be proposed by conventional methods (Fig. 1).
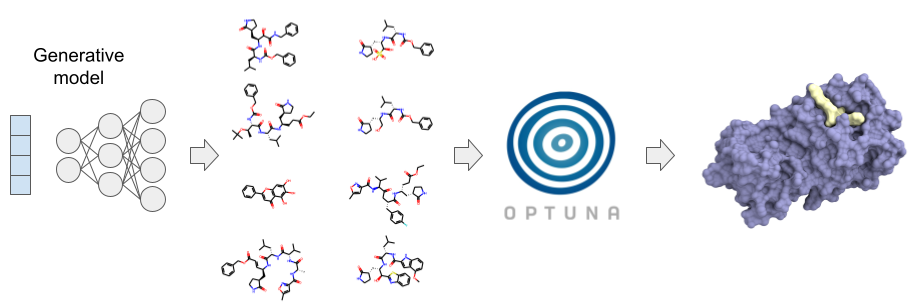
Figure 1: Conceptual diagram of PFN’s drug discovery platform.
Designing SARS-CoV-2 Mpro Inhibitor
Previous studies by Professor Akaji have revealed that the peptidic compounds (left) and non-peptide-like compounds (right) shown in Fig. 2 inhibit the SARS virus Mpro [1]. In this joint research, we utilized the PFN platform for molecular design and modeling in virtual space using AI to discover simple and non-peptide-like compounds that inhibit SARS-CoV-2 Mpro. As a result of synthesizing 13 compounds from the proposed compounds in the real world and conducting activity tests, 7 compounds showed inhibitory activity on Mpro of SARS-CoV-2, and are thus found to be promising lead compounds for the search for new coronavirus therapeutic agents. Although further optimization based on the lead compounds is needed to identify a therapeutic drug, this result indicates that PFN’s AI drug discovery platform can be applied to drug discovery in the real world.
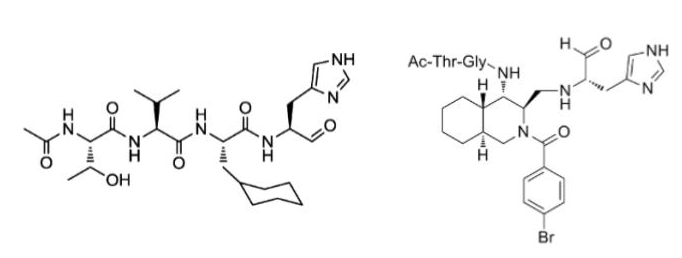
Figure 2. Structures of peptide compound (left, reference [2]) and non-peptide compound (right, reference [3]).
Conclusion
This study is significant in the sense that it provided a new approach to the development of COVID-19 therapeutic agents, as well that it works for drug discovery targeting cysteine proteases such as cathepsins, caspases, and also hopping of peptide scaffold. PFN will continue to improve AI drug discovery technology, seek partners who will jointly work on the AI-driven drug discovery to accelerate research toward practical application.
References
[1] Akaji, K. YAKUGAKU ZASSHI 2021, 141, 215-233.
[2] Akaji, K. et al. J. Med. Chem. 2011, 54, 7962-7973.
[3] Ohnishi, K. et al. Bioorg. Med. Chem. 2019, 27, 425-435.
Tag