Blog
2022.05.30
材料探索のためのユニバーサルなニューラルネットワークポテンシャル
So Takamoto
Head of Research Department, Materials & Drug Discovery Division

PFNリサーチャーの高本です。この投稿は、2022年5月30日にNature Communicationsで出版された論文 ”Towards Universal Neural Network Potential for Material Discovery Applicable to Arbitrary Combination of 45 Elements” [1] の解説記事です。論文はオープンアクセスで、以下のURLから閲覧可能です: https://www.nature.com/articles/s41467-022-30687-9
この論文で紹介されているPFPと呼ばれる技術は、PFNとENEOS株式会社が共同出資により設立したPreferred Computational Chemistryが提供している汎用原子シミュレーションソフトウェアMatlantis™においてコア技術として用いられています。なお、この研究成果はPFNとENEOS株式会社との共同研究によるものです。
要約
- 材料探索のために、未知の構造を扱える高速な原子シミュレーション技術が望まれていた。
- 汎用的な利用を目標とした新しいデータセットの構築と独自のニューラルネットワークアーキテクチャの設計をPFNのスーパーコンピュータを活用して行い、深層学習による原子シミュレーション技術PFPを開発した。
- PFPを材料探索を含む実際の多様な応用例に適用し、定量的に良い性能を持つことを示した。
背景
材料探索と原子シミュレーション
材料開発は科学技術の発展と深い関係にあります。例えば合金や樹脂の活用、窒素固定、半導体技術の進歩など、科学技術の発展はしばしば材料開発と一体のものでした。この関係は脱炭素や再生可能エネルギーなどの今後期待される技術に対しても同様で、材料開発は引き続き社会の発展の技術的な原動力として位置づけられています。
私達を含むこの世界にある様々な物質は、たかだか100種類程度の元素からなる原子の集まりによって作られています。また、物質の持つ多様な性質の起源をたどると、しばしば原子レベルでの振る舞いとして説明することができます。そこで、物質の性質を原子レベルでの振る舞いから説明しようとする試みが活発に行われてきました。近年では特にコンピュータの発達によって、物理シミュレーションを利用した材料開発が広く行われるようになってきています。材料の候補として理論上考えられる数は極めて膨大で、人類が今までに見つけられたものはそのごく一部であると考えられています。そのため、コンピュータの中で材料探索を行うことができれば、今後の社会において非常に魅力的なツールとなる可能性を秘めています。
このように世の中を物理法則とシミュレーションによって記述することを目指す際に長年の課題となってきたのは、低コストかつ汎用性のある近似モデルの構築です。原子レベルの現象を記述する量子力学を近似せずに直接計算することには極めて非現実的な計算時間がかかってしまうため、量子化学計算と総称される様々な近似を導入したシミュレーション技法が提案されてきました。密度汎関数理論(DFT)はコストと精度の両立に成功した量子化学計算手法の一つです。しかしながら、実世界で注目される現象には少なくともナノ秒オーダーの時間、ナノメートル以上のスケールが要求されることも多く、シミュレーションでは1回の計算でも100万ステップ、1000原子程度のスケールが必要になります。こういった例では計算可能なスケールと再現したいスケールとの間にまだ何桁もの開きがあるのが現状でした。
ニューラルネットワークポテンシャル
このため、量子化学計算をバイパスして、計算結果を直接推定する計算モデルが考えられてきました。特に原子の運動を再現することを目的としてエネルギー曲面を推定する計算モデルをポテンシャル(原子間ポテンシャル、力場)と呼び、従来より個別の材料に適用可能な経験的ポテンシャルが多数考えられてきました。また近年では深層学習の発展とともに、ニューラルネットワークによってポテンシャルを計算するニューラルネットワークポテンシャル(NNP)が注目され、深層学習の一分野として、またマテリアルズ・インフォマティクスの一分野として活発な研究開発が行われています。
NNPは、他のNNの応用領域と比較してユニークな特徴が数多くあります。例えば、入力されるデータは3次元空間に埋め込まれた大きさが不定のグラフで、近接するノード間の3次元的な位置関係が注目されます。推論するものはエネルギーなのでスカラー値の回帰問題となりますが、それだけでなく出力の微分値も推論時に利用されます(エネルギーの座標微分、つまり原子に働く力に相当します)。また、推論モデルを直接使うのは通常は人間ではなく物理シミュレータで、シミュレーションのために入力を少しずつ変えながら何度も繰り返し推論するなどの操作が頻繁に行われます。そのこともあり、シミュレータを動かすために前提条件となる性質、例えば回転や平行移動などの操作によってエネルギーは不変、あるいはエネルギーは離散的な変化をしないなどの様々な物理的制約を満たすことが望まれています。こういった条件を満たしたNNPのための深層学習アーキテクチャが提案されており、また大幅な精度向上が報告されています。
NNPを利用した材料シミュレーションは近年では基礎研究の段階を超え、応用を目指して幅広い領域で注目が集まっています。2020年にはFacebook AI Researchとカーネギーメロン大学が再生可能エネルギー貯蔵のための触媒開発を目的としたAIの開発を進めるOpen Catalyst Project [2]を開始しています。スーパーコンピュータ界のノーベル賞とも言われるゴードンベル賞で同じく2020年に受賞したトピックもまたNNPの大規模並列計算にまつわるものでした。[3]
課題
しかしながら、これらのイノベーションを迎えても、材料探索を行う上ではなお大きな課題が残っていました。それは未知の材料を扱う汎化性についての問題です。
今まで、ポテンシャルは特定の物質、特定の現象を再現するものとして開発されるのが通常でした。これは、十分な精度を確保するためには特定の原子構造をターゲットとしてポテンシャルを開発しなければいけないと考えられていたためです。
このことはNNPにおいても同様で、既存のデータセットもすべて特定のドメインに絞って作成されてきました。関連して、新しいデータセットが登場するとそれに対して有効なNNPアーキテクチャを考えるということが繰り返されてきました。これを自動運転車に例えるなら、特定の街や通りごとのデータセットがあり、その街や通りに対する自動運転車AIを開発するというアナロジーを考えることができます。このことはデータセットが限られている中で精度の良い推論モデルを作ることを考えると合理的な方法ではありました。
しかしながら、材料探索というタスクを考えると、未知の材料や仮想的な材料に対して良く動作することが要求されます。例えば新しい仮想的な材料を考えたときに、そのような材料が現実に存在できるのかどうか、また想定したような化学反応は起きるのかどうかといったことが考察の対象となります。つまり、既知の材料を扱えるだけでは性能が不十分ということを意味しています。材料の世界は機能性分子の形に限定しても10の60乗個の候補があると言われており、現象を再現するための複合的な構造ともなればそれを遥かに上回る組み合わせとなります。そのため、すべての構造を網羅するデータセットを作ることは非現実的です。先の自動運転車の例でいうなら、街が際限なく広がっているので知らない街を運転できるようにしたい、という要望に例えることができます。
それでは、汎用的なポテンシャルというものは作ることができないのでしょうか。あらためて解くべき問題を考えてみると、原子の世界では登場人物は元素であり、その種類は有限個です。しかもその相互作用は予測不可能な突拍子もないものではなく、シュレディンガー方程式という単一の方程式によって良く記述できているものでした。また、近年のNNの研究をみると、いくつかのポジティブな例を見つけることができます。例えば自然言語処理の分野では汎用的な言語モデルが登場し、その性能の高さから大いに注目を集めました。このように、いくつかの分野ではある種の汎用性の獲得が実際に実現しつつあります。
このことから、もしも適切なデータセットとNNPアーキテクチャ、そして十分な計算資源が用意できれば、一般的な原子の相互作用の内部表現を持つ「ユニバーサルなNNP」を得ることは必ずしも不可能ではないのではと考えました。そこで私達はこの意欲的な課題を目標とし、解決するべき課題を洗い出し、要素技術の開発を進めてきました。
手法
NNの技術開発は、データセットとアーキテクチャの両面が重要です。私達はこの両方について技術開発を進めてきました。なお、成果として得られたNNPにはPFPという名前をつけています。
データセットは、不安定な構造を積極的に集め、できるだけ多様なパターンが含まれるように構築しています。また、知られている材料のデータベースなどに限定するのではなく、とある原子構造に対してすべての元素の組み合わせを当てはめて計算するなど、積極的に仮想的な構造が含まれるように取得しています。これは、既知の安定した原子構造を集める従来のデータセットとは大きく異なる方針です。具体的には、様々な結晶や分子構造で元素が不規則に置換された構造、様々な異なる元素が同時に存在する乱れた構造、温度や密度を様々に変化させた構造などが含まれるようにしています。元素の種類は論文投稿時点では45種類を対象としています。
このような新奇なデータセットを1から構築するには、相応の計算資源が必要でした。データセットにおける教師データはDFT計算となります。上記のようにDFT計算は大きな計算コストがかかることが問題でしたが、計算対象を適切なサイズにすることに加え、PFNのクラスタ(MN-1およびMN-2)にあるGPUを1枚換算で合計164年分以上使うことで、ある程度の大きさのデータセットを作成しました。
実際の開発では、NNPアーキテクチャの開発や実際のシミュレーションへの適用を通して、できるだけ汎用性が確保できるよう、データセットの構築の手順に繰り返しフィードバックを行いながら開発を進めています。このため、研究が進んでデータセットが高度化されるほど開発の難易度が上がっていくという意欲的な取り組みとなっています。この取り組みはPFNエンジニアの品川が主体となって進めました。(なお、論文投稿以降も引き続きデータセット構築を続けており、現時点ではGPU換算で1000年分を超えるまでになっています。)
NNPアーキテクチャもこのような乱れた構造を破綻なく扱える設計にする必要があります。アーキテクチャとしては、PFNリサーチャーの高本が考案した独自のグラフニューラルネットワーク(GNN)のTeaNet [4]をベースとして、さらに改良を加えたものを使用しています。これは経験的ポテンシャルに組み込まれてきた物理的なモデルとGNNを融合したアーキテクチャで、グラフの中に2階のテンソル量を流すモデルとなっています。局所的な位置関係の情報を捉えつつ、剛体回転や剛体移動などに対して現象が変わらないという物理的な要請をモデルとして持つことを可能にしています。
データセットとアーキテクチャ開発はまさに深層学習の両輪で、両方の技術の発展によってできることも増えていきました。開発の途中からは学習して得られたNNPを利用して原子シミュレーションを行い、追加のデータセットを取得することも可能となりました。
このように作成したデータセットから一部を抽出したものをHigh-temperature multi-elementデータセット(HME21)として公開しています(https://doi.org/10.6084/m9.figshare.19658538)。先述のように、本研究では汎用性を目的として非常に乱雑な原子配置を持たせたデータセットを構築しており、既存のNNP向けのデータセットと比較して難易度の高いものとなっていると考えられます。
また、NNPアーキテクチャ単体の性能評価として、HME21データセットに対するNNPアーキテクチャのベンチマーク結果を論文に載せています。ベンチマークにはオリジナルのTeaNetを使ったものを載せています。他NNPアーキテクチャと比較して高い性能を達成していることがわかります。
HME21データセットの公開には大きく3つの目的があり、1つ目は論文内に記述のある汎用的なNNPのためのデータセットの作成方法についての補足情報、2つ目はPFPのアーキテクチャの性能を示すためのベンチマーク用データセット、そして3つ目は今後のNNP開発におけるデータセットとして使われることです。HME21データセットをはじめとした本研究の成果物を通して、汎用的なNNPに関連した学術領域がさらに発展することを期待しています。
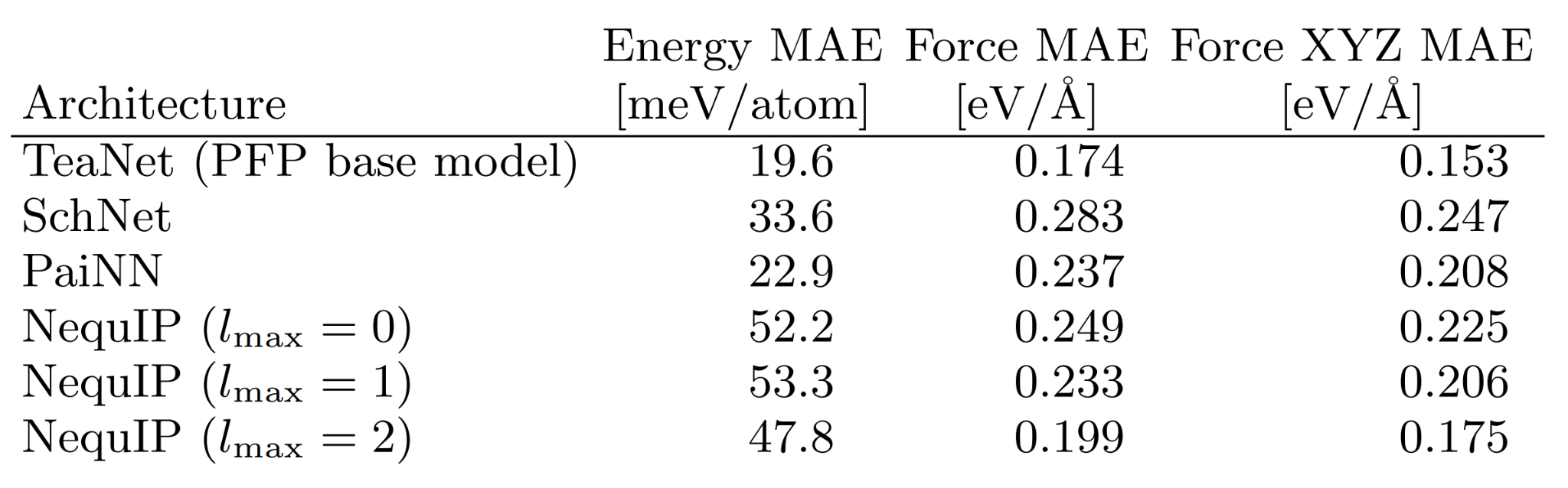
Benchmark performance of NNP for the force and energy prediction for the HME21 dataset. Excerpt from [1].
結果: PFPを利用した材料シミュレーション
作成したPFPを実際の材料シミュレーションの例に適用しました。背景に書いたように、私達の目的は材料探索なので、単にデータセットへの適合度を見るだけでは不十分で、現実の問題に対して良い性能が出ているかどうかを調べる必要があります。
今回の論文では、リチウムイオン電池のLiイオンの拡散挙動、金属有機構造体の分子吸着挙動、金-銅合金の相転移現象、そして炭化水素合成を行うフィッシャー・トロプシュ反応の再現および触媒の材料探索という、材料も対象とする現象も互いに大きく異なる4つの例を題材として原子シミュレーションを行いました。論文のAuthor contributionを見ていただければわかるように、プロジェクトの多くのメンバーがPFPを利用した材料シミュレーションに携わっています。論文に収録された内容からは外れますが、この4つの適用例以外にも様々な例への適用が試みられています。公開可能ないくつかの例をMatlantisのWebページにも載せていますので、興味のある方はぜひご覧ください。
以下では、4つの例を個別に紹介していきます。ただし個別の事例に深入りして説明すると非常に長くなってしまうため、詳細な計算手法や計算結果についてのディスカッションは論文本文を読んでいただくことにして、ここではそれぞれの例で対象とした材料の説明や、シミュレーションを行う際の技術的特徴にしぼって簡単に紹介させていただきたいと思います。
リチウムイオン電池のリチウム拡散
電気自動車や携帯機器などをはじめとしてリチウムイオン電池の重要性は日に日に高まっていますが、リチウムイオン電池の重要な特性として充放電速度があります。これに関わっているのが材料中のリチウム原子の拡散のしやすさで、活性化エネルギーという量が注目されます。
対象とする現象は、バルク結晶中の格子間原子の振る舞いを再現する問題になります。特に、拡散現象は経路の途中のエネルギーが最も高くなる点(鞍点)によって支配されるので、そういったエネルギーの不安定な構造を正しく再現できなければいけません。計算手法としては、Nudged Elastic Band (NEB)法および補助的に分子動力学(MD)計算を行って検証しています。
今回の適用例では、3つの異なる拡散パスに対して活性化エネルギーを調べています。いずれも先行研究のDFT計算の結果を良く再現するもので、良好な性能が出ています。
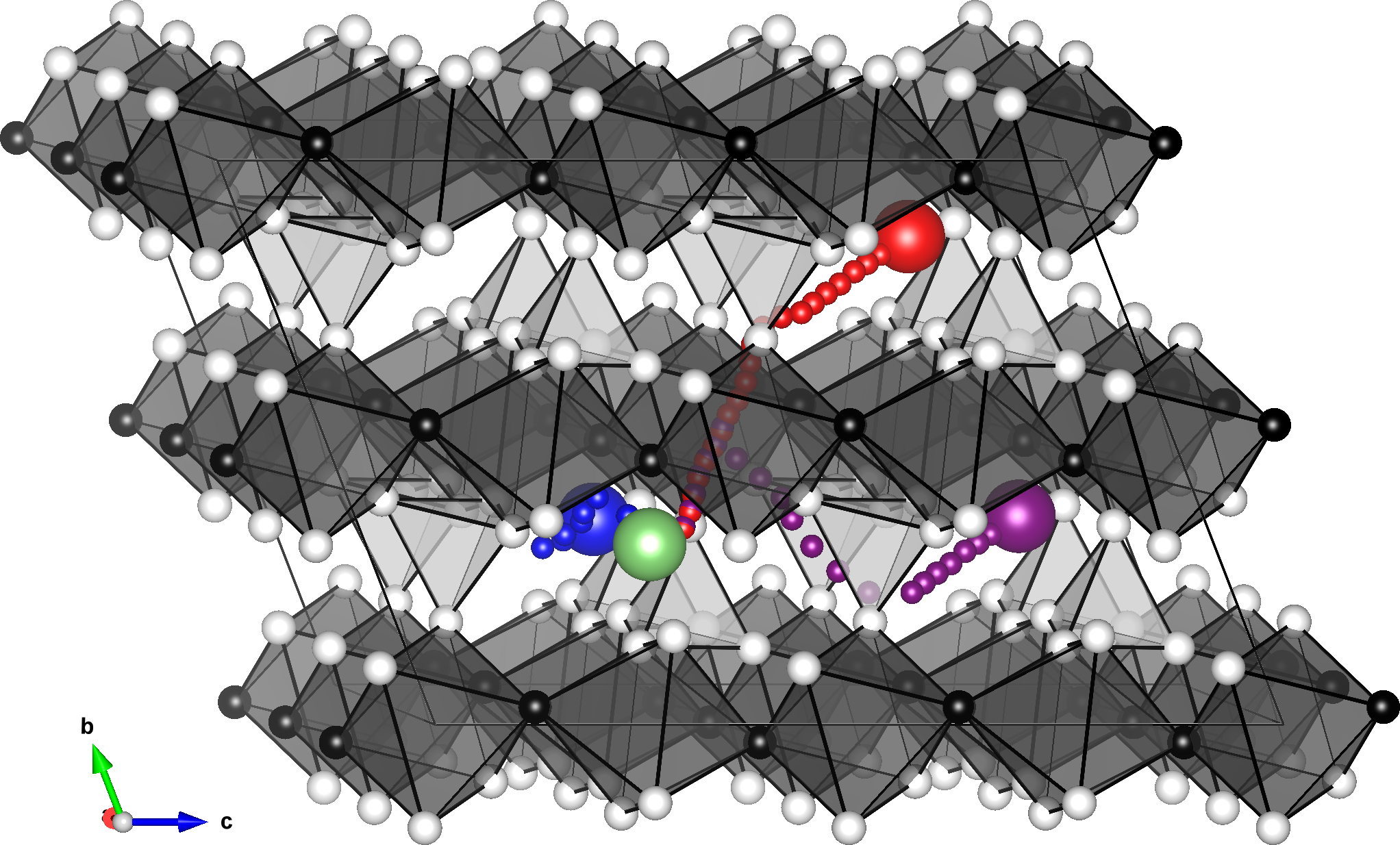
Lithium diffusion paths. Excerpt from [1].
金属有機構造体の分子吸着挙動
有機金属構造体(Metal-organic framework, MOF)は、極めて高い表面積を持つナノ多孔体材料の一種です。様々な分子を利用することで人工的に細孔構造を制御することができます。一部の金属原子では活性な不飽和サイトが小分子の吸着サイトや触媒として作用する場合があります。
対象とする現象は、MOF構造の再現および水分子の吸着エネルギーです。MOF構造は有機物の構造と金属元素を含む複雑な化学構造をしているため、多様な元素が含まれる構造を精度良く再現する必要があり、従来のポテンシャルの活用が難しいものでした。また、金属の吸着サイトへの水分子の吸着エネルギーを調べています。この計算では、計算結果にD3補正という追加のファンデルワールス相互作用の補正を組み込んだ比較も行っています。
今回の適用例では、8種類のMOF構造および5種類の吸着エネルギーについて評価を行っています。いずれも平均誤差が数%の範囲内で推定できています。
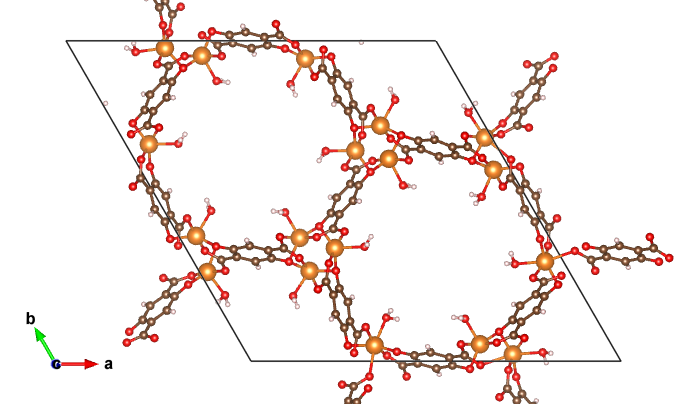
MOF structure illustration. Excerpt from [1].
金-銅合金の相転移現象
金-銅合金は、COやアルコールの酸化触媒として研究されてきた材料です。金-銅合金は幅広い組成域で固溶し、また秩序-無秩序構造の相転移が起きることが知られています。ミクロな原子の配置は触媒の性質と深く関わっており、現象解明に重要な要素です。
対象とする現象は、合金の秩序-無秩序構造の相転移です。合金自体の再現のために金属結合を再現する必要があることに加え、配置の乱れによるエネルギーの変化を精度良く再現できる必要があります。また、計算手法はメトロポリスモンテカルロ法を使っています。
今回の適用例では、Cu3Au, CuAu, CuAu3の3種類の組成について相転移の温度を調べています。CuAuで最も相転移の温度が高くなるという実験結果と同じ振る舞いが算出されています。
フィッシャー・トロプシュ反応と触媒の材料探索
フィッシャー・トロプシュ(FT)反応は、触媒を使って水素と一酸化炭素から炭化水素を合成する反応です。この反応は、持続可能なエネルギー源から石油の代替物質を作り出すテクノロジーとして注目されています。また、今回の例では実際に材料探索を行い、有望な添加元素を探し出すところまでを行いました。
対象とする現象は、コバルト表面でのメタン化反応と一酸化炭素の解離過程に注目します。これは、金属表面と有機物、化学反応の過程、また表面吸着の効果というように多くの要素が絡み合った現象です。今回は20個の素反応について、DFTを使った先行研究と比較しています。また、元素を置換しての触媒構造探索を通して、PFPが材料探索に使えるかどうかのベンチマークを行っています。
今回の適用例では、活性化エネルギーを平均絶対誤差0.1 eV未満という良い精度で再現できています。さらに、材料探索ではバナジウムの添加が有望であることが示唆されましたが、これに関して文献をあたると実際にバナジウムの添加が良いとされる実験の報告があることがわかりました。
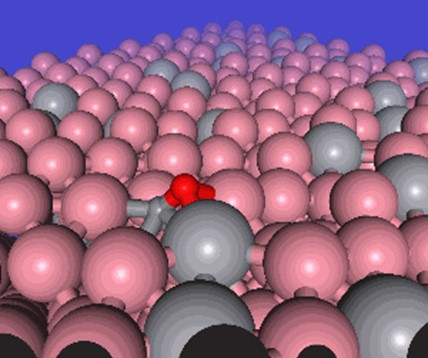
CO adsorbed configuration of a Co surface with V promoters. Excerpt from [1].
ディスカッション、その他トピック
上記の例では、いずれもシミュレーションが動いただけでなく、定量的な議論のできる結果が得られています。特に、データセット構築の際にこれらの材料を明示的に入れていないにもかかわらず再現できたこと、またこれらの4つの例すべてを単一のモデルを使って再現可能であったことは印象的な結果となりました。
今回紹介したものは結果の定量的な比較のために先行研究でDFT計算などが行われているものを例にあげています。先行研究では多大な計算資源が投入されて解かれていたものが、今回のPFPを利用した計算ではいずれも高速に解けていることは注目に値します。大規模な計算が可能になることによりシミュレーションの自由度を上げることができ、また幅広い材料探索を行うことができることが期待されます。また、研究者にとってはインタラクティブな研究活動を通して仮説検証を高速に繰り返すことができるようになります。
計算時間の比較として、白金3000原子の系を例にあげています。DFTでは1つの構造を計算するのに推定で2ヶ月必要な計算が、PFPを使った場合には0.3秒で計算が終わりました。これはPFPによって1000万倍オーダーで速くなったことに相当しており、圧倒的な高速化となっています。
今回の紹介記事では取り上げきれませんでしたが、論文中では他にもNNの学習に関わるトピックが含まれています。例えば、物理シミュレーションの近似の都合で互いに完全にコンシステントではない複数種類のデータセットを利用してNNPの学習を行っているのですが、これによってデータセットAにしか含まれていない元素がデータセットBに対する推論でもおおむね動作するようになるなど、汎化性能の観点から興味深い振る舞いがみられました。
今後に向けて
PFPをコア技術として、私達は材料探索を行うためのSaaS製品Matlantisを開発しています。Matlantisでは実際にPFPを利用した原子シミュレーションが実行可能な環境を提供しています。
論文投稿以降も私達は積極的に研究開発を続けています。論文のモデル以降、MatlantisではPFPは2回のメジャーアップデートが行われ、今ではDFT計算に基づくデータセットの大きさは2200万構造、用いた計算リソースはGPU 1000年分を超えており、さらなる精度向上と適用範囲の拡大を続けています。より幅広い材料探索のための計算速度向上とスケールの向上も重要な課題となっています。
PFPを活用した具体的な材料探索への取り組みも進めています。また、NNP以外での計算材料科学と深層学習の融合も研究を続けています。分子動力学よりもスケールの小さい側、大きい側のいずれにも工学的に重要な問題が存在しており、NNPとは別の、あるいはNNPと組み合わせての機械学習技術の適用について研究を進めています。このような活動内容に興味を持たれた方はぜひお問い合わせいただければと思います。
関連して、分子動力学のスケールでの技術開発も進めています。汎用的なNNPを利用可能になったということは、材料の大海原を探検する上での地図を手に入れたようなものです。これは確かに単独でも有用な技術ではありますが、この地図を使って広大な材料の世界を俯瞰する技術を洗練させていくことで、材料探索の地平はさらに広がっていくと考えられます。機械学習と物理シミュレーションの融合は今後も続いていくように思われます。
最後になりますが、PFNでは材料科学分野で一緒に働いていただけるメンバーを募集しています。Matlantisのサービス開発から要素技術開発、材料探索にいたるまで様々な面での携わり方がありますので、興味があればぜひご連絡いただければと思います。
外部リンク
- PC Watch (2021/07/07): 3千CPUで数カ月かかる計算が0.1秒で完了。汎用原子レベルシミュレータ「Matlantis」~PFNとENEOSがクラウドサービスで提供開始
- 日経電子版 (2021/08/04): 専門家も脱帽 深層学習を使った量子化学計算の威力
- メガソーラービジネス (2021/08/17): 内燃機関「カーボンゼロ」へ、期待の再エネ合成燃料
- 日経電子版 (2021/08/19) ENEOS執行役員「合成燃料、触媒で歩留まり改善」
- 日本の研究.com (2021/09/28) 【PR】第一原理計算結果を数秒で返す汎用原子レベルシミュレータMatlantis™ 開発インサイドストーリー
- テンミニッツTV (2022/01/01): 2022年は希望の年、「課題解決先進国」日本への動きが活発化 小宮山宏
- 日経電子版 (2022/02/05): プリファード、化学計算3日を30分に 材料探しに威力
使用事例
- Matlantis case study, “高速な汎用原子レベルシミュレータが触媒開発に変革をもたらす”, https://matlantis.com/ja/case-study/case1
References
[1] So Takamoto, Chikashi Shinagawa, Daisuke Motoki, Kosuke Nakago, Wenwen Li, Iori Kurata, Taku Watanabe, Yoshihiro Yayama, Hiroki Iriguchi, Yusuke Asano, Tasuku Onodera, Takafumi Ishii, Takao Kudo, Hideki Ono, Ryohto Sawada, Ryuichiro Ishitani, Marc Ong, Taiki Yamaguchi, Toshiki Kataoka, Akihide Hayashi, Nontawat Charoenphakdee, and Takeshi Ibuka “Towards universal neural network potential for material discovery applicable to arbitrary combination of 45 elements” Nature Communications, 13, 2991 (2022). https://doi.org/10.1038/s41467-022-30687-9 CC-BY 4.0 (http://creativecommons.org/licenses/by/4.0/)
[2] Open Catalyst Project. https://opencatalystproject.org/
[3] https://www.acm.org/media-center/2020/november/gordon-bell-prize-2020
[4] So Takamoto, Satoshi Izumi, Ju Li “TeaNet: Universal neural network interatomic potential inspired by iterative electronic relaxations” Computational Materials Science, 207, 111280, DOI: 10.1016/j.commatsci.2022.111280 (2022).