Blog
創薬の初期段階として、薬の候補となるヒット化合物の探索は重要なプロセスのひとつです。実際のタンパク質と化合物ライブラリーを用いてウェットのスクリーニング実験を行い、ヒット化合物を同定することも可能ですが、膨大な実験に伴う試薬代・人件費・時間的コストなどを軽減するために、コンピューターを利用して薬の候補を絞り込むバーチャルスクリーニングと呼ばれる手法が重要となっています。バーチャルスクリーニングは主に、化合物のみを用いるリガンドベースの手法と、タンパク質の立体構造と化合物構造の両方を用いる構造ベースの手法、さらにそのほかの手法に大別することができます。このうち構造ベースの手法では、タンパク質の立体構造と化合物から、高速に結合結合親和性と結合ポーズを予測する必要がありますが、通常これにはドッキングシミュレーション[1]が用いられています。数百万~数千万という膨大な数の化合物に対するスクリーニングを高速に実行するために、これまでスパコンなどの有する多数のCPUを用いて、並列にドッキングシミュレーションを行い、スクリーニングを加速することが行われてきました[2]。一方、GPUを用いた並列化例も、近年いくつか報告されています[3]。特にPFNではMN-2 [4]などGPUを多数搭載した計算機が使用可能なため、GPUを用いた計算の高速化は重要です。本記事では、弊社におけるGPUを用いたドッキングシミュレーションの実装と高速化の事例について紹介いたします。
方法と実装
ドッキングシミュレーションの方法
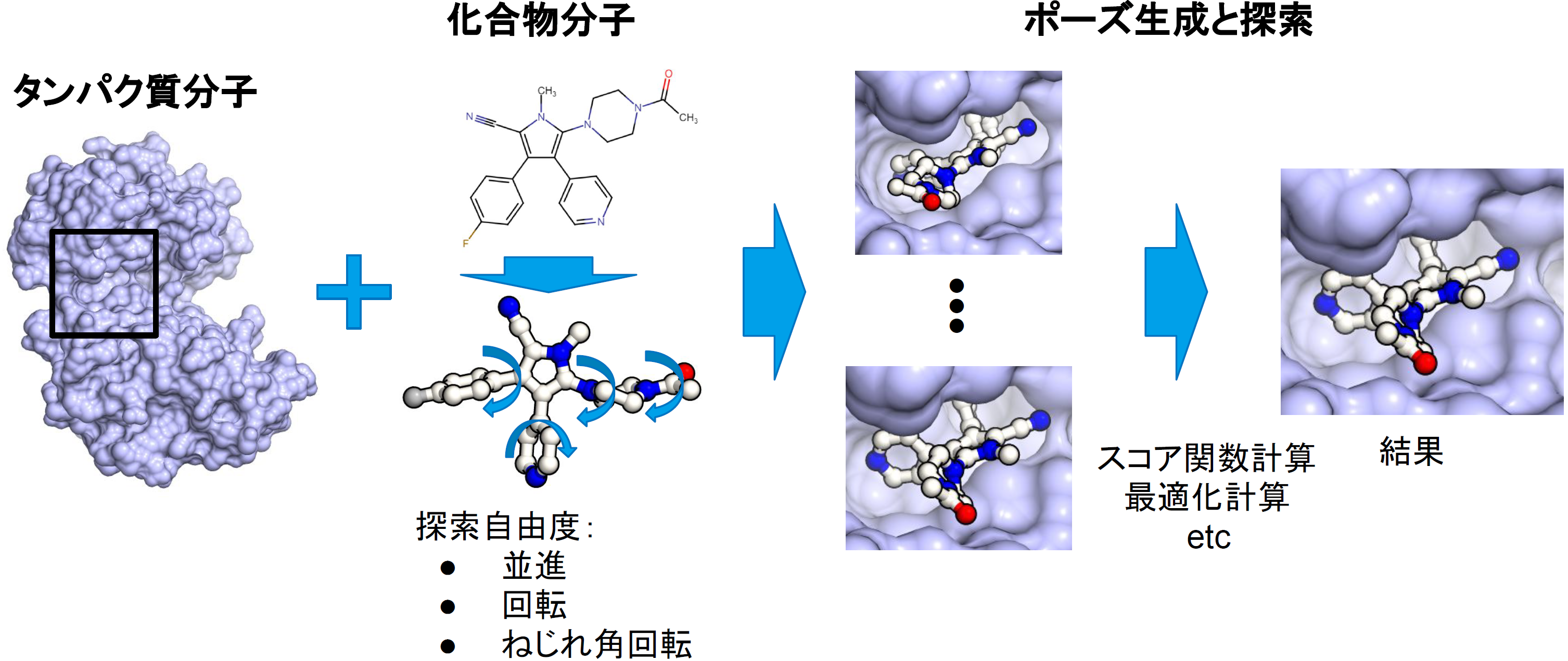
図1:ドッキングシミュレーションの方法概要
ドッキングシミュレーションの方法には多数手法が提案されていますが[1, 5]、ここでは、Autodock Vinaで用いられている方法[6]を採用しました。以下簡単に計算法の概略を説明します(図1)。まずこの方法では、リガンド(化合物)は回転可能な共有結合(rotatable bond)が回転し構造変化しうると考えますが、レセプター(タンパク質)側は構造変化しないという仮定で計算を行います。
すなわち、リガンドの配座(座標)は、全体の位置と向き、各rotatable bondの回転角度で表すことができます。ここでは、これらの自由度のセットを内部座標と呼ぶことにします(それに対し、各原子の座標をxyzで表現したセットをデカルト座標と呼ぶことにします)。これらの自由度を探索して、最もスコア関数が良くなる配座を探すことで、レセプターのポケットに最も適合するリガンドの配座を得て、その配座からさらにレセプターとリガンド間の結合親和性(結合自由エネルギー変化ΔG)を予測します。
この探索を行うための大まかな手順は以下のとおりです。まず、上述の内部座標の各自由度について何らかの方法でサンプリングを行います(Autodock Vinaの場合は、温度一定のモンテカルロサンプリングを行っている)。次に、その座標値から、BFGS法[7]を用いたエネルギー最小化を行い、スコア値と座標値を局所最適解へと更新します。そして得られたスコア値(エネルギー値)をもちいて、メトロポリス判定を行います。
ここでは、Autodock Vina (とその派生ソフトウェア)で用いられている方法を紹介しましたが、他には、勾配計算が必要なBFGS法による最適化を行わないものや、モンテカルロ探索以外の探索法(進化的アルゴリズムや群知能などのメタヒューリスティクスアルゴリズム)を用いるものなど、これ以外にも様々な方法が提案されています[5]。
GPUによる高速化
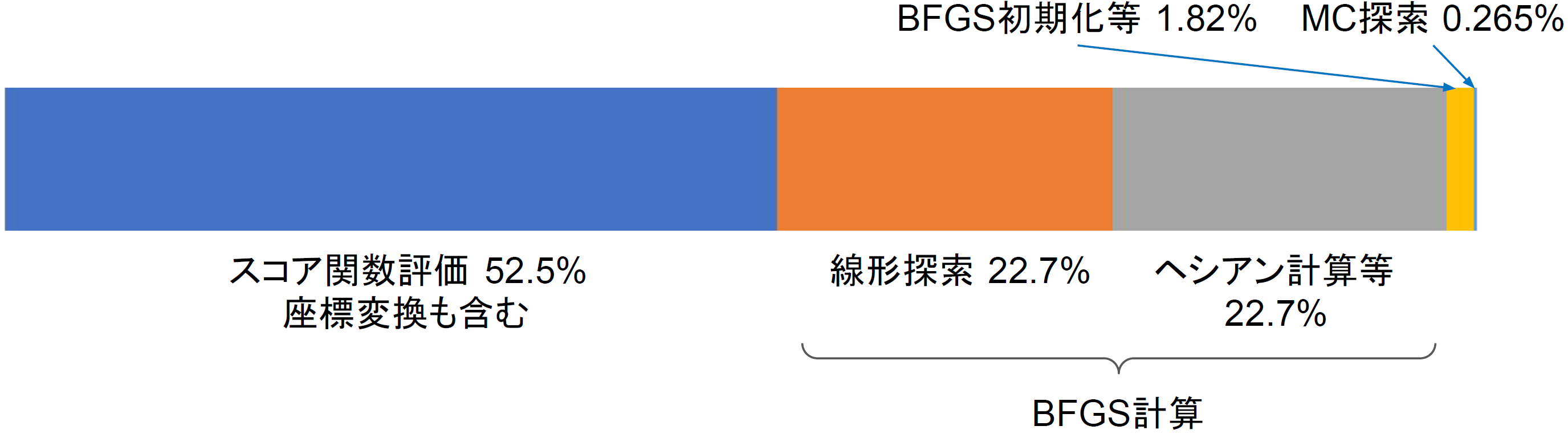
図2:CPU計算時の各所要時間
これらの手順のうち、計算量の大きい部分は、スコア値とその勾配計算を含むBFGS法による最適化の部分となります(図2)。この計算には
- 内部座標値からデカルト座標値への変換
- デカルト座標値を用いた、分子内相互作用計算
- デカルト座標値を用いた、タンパク・リガンド相互作用計算
- デカルト座標系の力(勾配)を内部座標系に変換
などの計算が含まれます。ここでは、これらの計算をGPUで並列化することにより、高速化を試みました。例えば、リガンド・レセプター間やリガンド内相互作用エネルギー(スコア)の計算は、原子ごとに計算をGPU threadに割り振り、並列化することが可能です。
また、バーチャルスクリーニングでは多数の分子を同一ターゲットタンパクに対してドッキング計算しますが、各化合物分子は並列して計算が可能です。またさらに、通常、探索効率を上げるために、1分子あたり複数回の探索を行う場合が多いですが、これも並列化が可能です。一方で、モンテカルロ計算部分(乱数の発生やメトロポリス判定部分など)は、計算量が少なく全体計算時間に占める割合が少ないため(図2)、並列化の効果が薄いと考え、CPUで行う実装としました。
ところで、このようなCPUとGPUでの計算を併用する実装では、単に逐次的に実装してしまうと、CPUで計算(モンテカルロ部分)を実行している際に、GPUが有効活用できないという問題があります。また、CPU・GPU間のデータ転送中もCPUが待機し、リソースが有効活用できないという問題もあります。そこで、分子群を2つのバッチに分け、各バッチを交互に計算を行う実装としました。すなわち、バッチ0がGPU計算を行っている間に、バッチ1のCPU計算を行う、等です。これにより、CPU、GPUの計算完了待ち時間軽減や、デバイス間データ転送のレイテンシを隠蔽できると期待されます。
実験と結果
計算条件
まず実験条件ですが、ドッキングポーズの評価スコア関数としては、vina score [6]を用いました。あるたんぱく質に対して、約1000万化合物からなるライブラリーをスクリーニングする問題設定でベンチマークを行いました(実際にはライブラリーからランダムに抽出した約50万化合物でパフォーマンス計測を行いました)。また、前処理として、化合物中のrotatable bond数(すなわち化合物の自由度)がなるべく同じになるものをグループ化し、3次元構造への埋め込み(embedding)を行いました。最終的なライブラリー中の化合物のrotatable bond数は0~20としました。そして、各分子について、16回ドッキングシミュレーションを行い、スコアの最良値を解としました。並列化の効果を検証するため、同時に64, 128, 256, 384分子のドッキングシミュレーション計算を行うケースを比較しました。計算環境としては、GPUにはNVIDIA Tesla V100 SXM2を、対照実験としてCPUにはIntel Xeon Gold 6254 CPU@ 3.10GHzを用いました。
結果
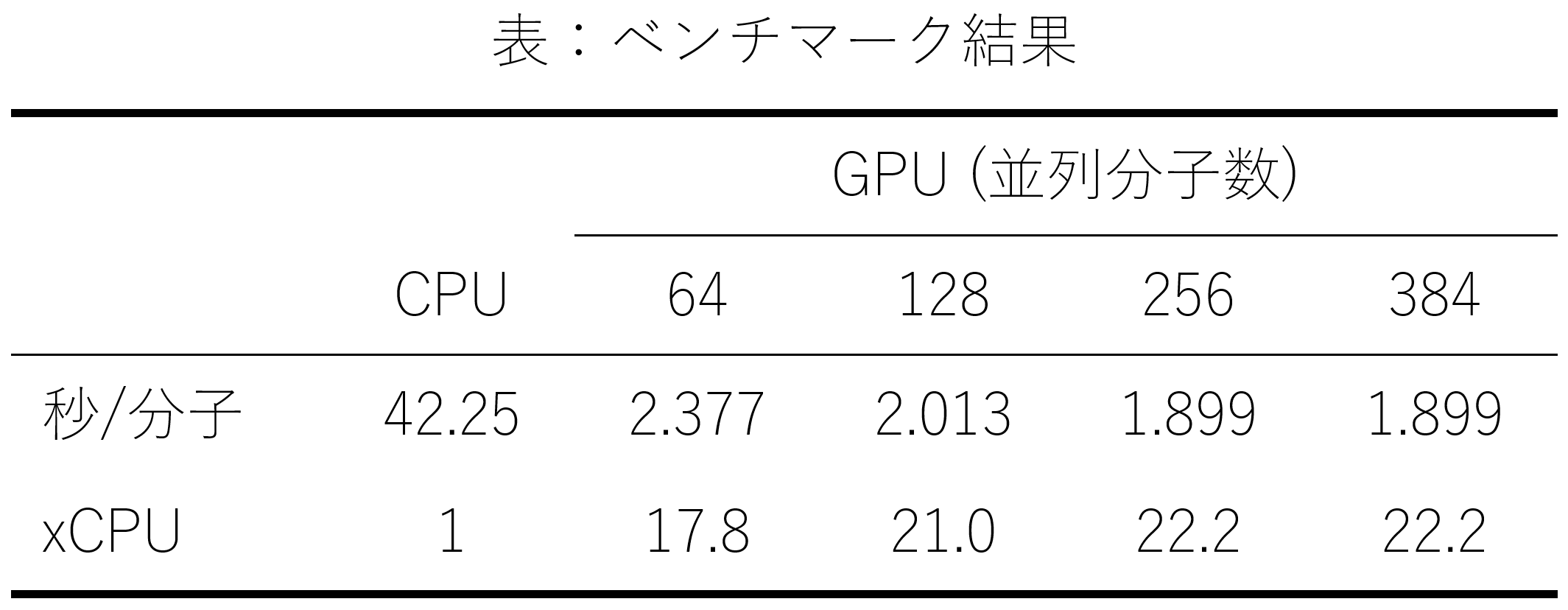
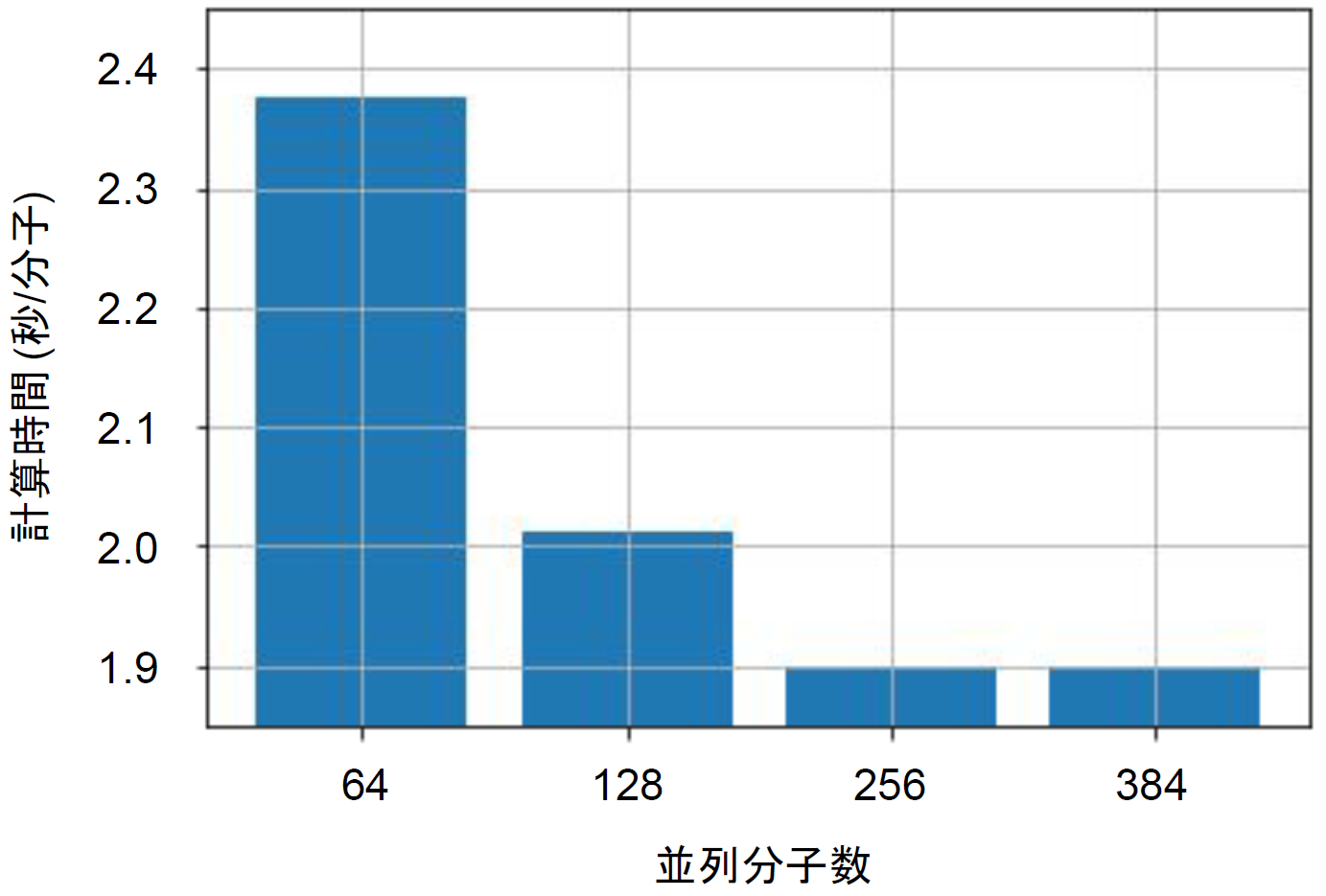
図3:ベンチマーク結果
各条件で、ドッキングシミュレーションに要する時間を、1分子当たりの平均としたものが図3です。この結果からは、V100 GPUを用いた場合は256分子程度までは、並列化による高速化の効果が現れていると考えられます。256分子で計算した場合、CPUに比べ平均で22.2倍の高速化になっていることが分かりました。これは、ベンチマークに用いた1000万化合物のライブラリーなら、MN-2搭載GPUの約1/5に相当する220台のGPUを用いれば1日弱でスクリーニングが完了する計算となります。
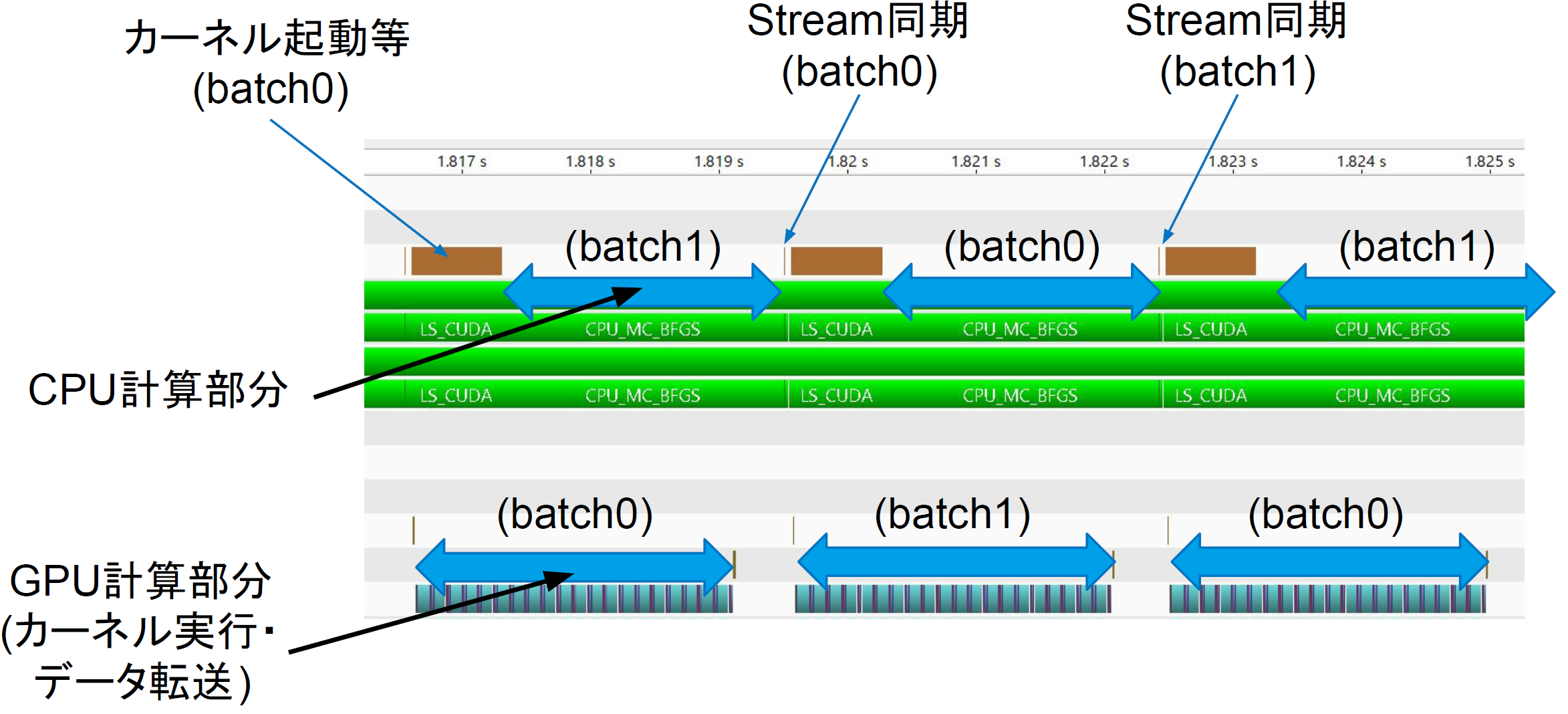
図4:プロファイリングとパフォーマンス計測
次に、バッチに分けて計算することで、CPU/GPUが効率的に使用されているかを検証しました。図に示すように、CPU/GPU間データ転送命令が発行された後、CPU側は別バッチのモンテカルロ計算を行うことで、データ転送のレイテンシを隠蔽することができていることがわかります。また、GPUで計算中にCPU側の計算を並行して行うことで、GPU側の計算終了待ち時間を有効活用できています。
まとめ
本稿では、弊社のインシリコ創薬の取り組みの一例として、バーチャルスクリーニングのためのドッキングシミュレーションの高速化の事例を紹介いたしました。一方で近年、深層学習を用いることで、従来法のような探索を行わずにドッキングポーズを出す方法も提案されるようになってきました [8]。今後、タンパク質の立体構造予測におけるAlphaFold2のように既存手法を置き換えるポテンシャルを持っていますが、現状では、従来法の活用も必要であると考えられます。弊社でも、従来法を用いたドッキングシミュレーションだけでなく、深層学習を用いたドッキング手法、さらにはインシリコ創薬・AI創薬の研究開発を進めています。このような研究開発に興味がある方は、下記の通り人材募集しておりますので、ぜひご応募ください。
参考文献
[1] 上原 彰太, 田中 成典 タンパク質-リガンドドッキングの現状と課題 日本化学会情報化学部会誌 2016 年 34 巻 1 号 p. 10-
[2] Dong D., Xu Z., Zhong W., Peng S. Parallelization of Molecular Docking: A Review. Curr. Top. Med. Chem. 18 1015-1028 (2018).
[3] Santos-Martins D., Solis-Vasquez L., Koch A., Forli S. Accelerating AutoDock4 with GPUs and Gradient-Based Local Search. J. Chem. Theory. Comput. 17 1060–1073 (2021)
[4] https://tech.preferred.jp/ja/blog/mn-2-is-up/
[5] Pagadala N. S., Syed K., Tuszynski J. Software for molecular docking: a review. Biophys. Rev. 9 91–102 (2017)
[6] Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading, J. Comp. Chem. 31 455-461 (2010)
[7] Nocedal J., Wright S.J. Numerical optimization. Berlin: Springer Series in Operations Research, Springer Verlag (1999)
[8] Stärk H., Ganea O.E., Pattanaik L., Barzilay R., Jaakkola T., EquiBind: Geometric Deep Learning for Drug Binding Structure Prediction. arXiv:2202.05146
Tag