Chemoinformatics / Materials Science
Discover new materials/drugs with computational science.
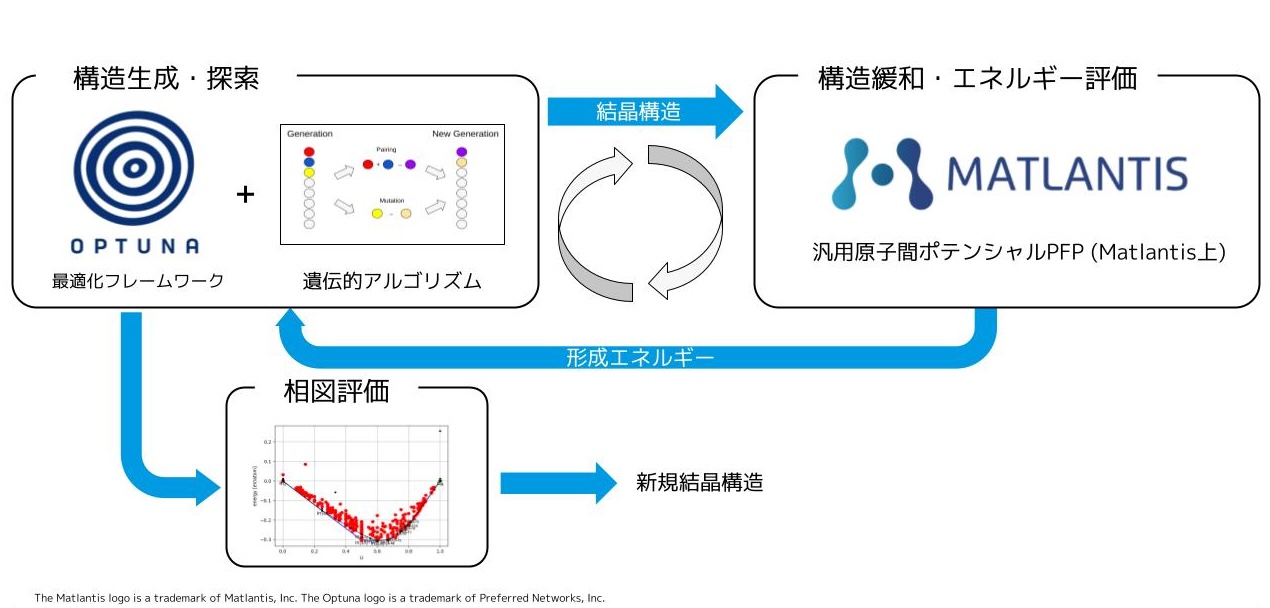
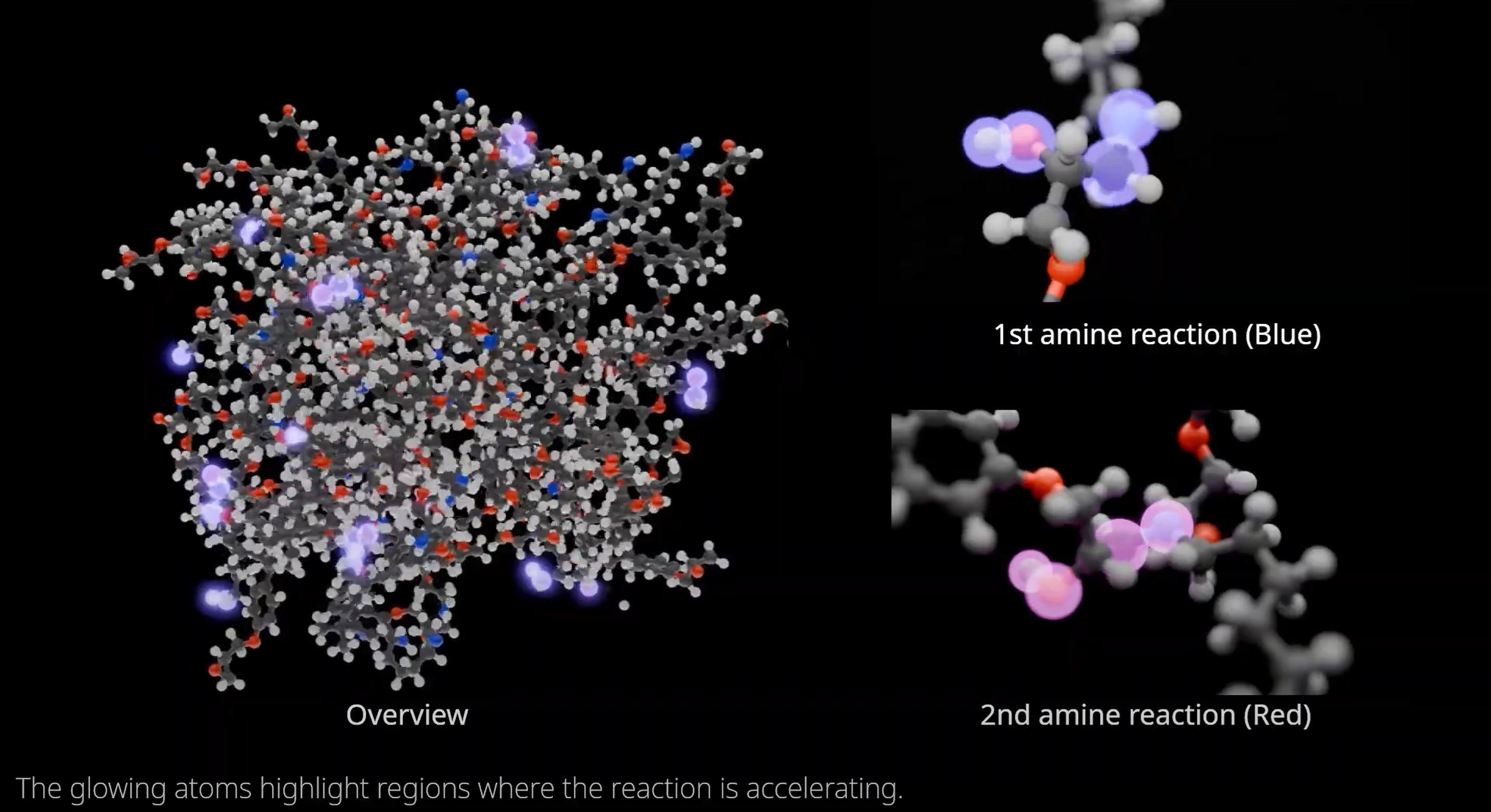
数値計算を用いた物性予想は、実験コストを削減し、新材料開発を促進する手段として注目されています。しかし、候補となりうる化合物や材料は非常に多く、また、材料を構成する電子、原子を1つずつ計算するには膨大なコストがかかるため、従来のシミュレーション技術で網羅的に扱うのは困難です。PFNでは深層学習と数値計算を融合させた物性予測システムを開発することで、より高速、高精度な物性予測を実現し、材料開発を促進することを目指しています。PFNではこのような技術を創薬・素材探索へ適用するための研究開発を行っています。
Publications
Efficient Universal Potential Distillation with Pre-trained Students in LightPFP
NeurIPS2025 AI4Mat
By : Wenwen Li, Nontawat Charoenphakdee, Yong-Bin Zhuang, Yuta Tsuboi, Ryuhei Okuno, So Takamoto
P-DRUM: Post-hoc Descriptor-based Residual Uncertainty Modeling for Machine Learning Potentials
NeurIPS2025 ML4PS
By : Shih-Peng Huang, Nontawat Charoenphakdee, Yuta Tsuboi, Yong-Bin Zhuang, Wenwen Li
A practical guide to machine learning interatomic potentials – Status and future
Current Opinion in Solid State and Materials Science
By : Ryan Jacobs, Dane Morgan, Siamak Attarian, Jun Meng, Chen Shen, Zhenghao Wu, Clare Yijia Xie, Julia H. Yang, Nongnuch Artrith, Ben Blaiszik, Gerbrand Ceder, , Kamal Choudhary, Gabor Csanyi, Ekin Dogus Cubuk, Bowen Deng, Ralf Drautz, Xiang Fu, Jonathan Godwin, Vasant Honavar, Olexandr Isayev, Anders Johansson, Boris Kozinsky, Stefano Martiniani, Shyue Ping Ong, Igor Poltavsky, KJ Schmidt, So Takamoto, Aidan P. Thompson, Julia Westermayr, Brandon M. Wood
npj Computational Materials
By : Zetian Mao, Wenwen Li, Jethro Tan